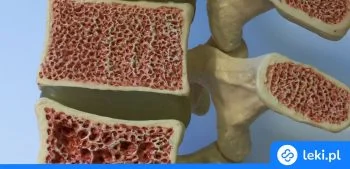
Teryparatyd jest nowoczesną substancją czynną stosowaną głównie w leczeniu osteoporozy u dorosłych, która pomaga odbudować gęstość kości i zmniejsza ryzyko złamań. Wyróżnia się skutecznością zarówno u kobiet po menopauzie, jak i u mężczyzn, a także u osób z osteoporozą wywołaną długotrwałym przyjmowaniem glikokortykosteroidów. Dostępny jest wyłącznie w postaci roztworu do wstrzykiwań, co pozwala na precyzyjne dawkowanie i szybkie działanie. Teryparatyd należy do grupy hormonów przytarczyc i działa poprzez pobudzanie naturalnych procesów budowy kości.
Jak działa teryparatyd?
Teryparatyd jest substancją czynną z grupy hormonów przytarczyc. Wpływa na procesy zachodzące w kościach, pobudzając ich odbudowę i zwiększając gęstość mineralną. Dzięki temu zmniejsza ryzyko złamań u osób dorosłych, zwłaszcza w przypadku osteoporozy1234567.
Dostępne postacie i dawki
- Roztwór do wstrzykiwań we wstrzykiwaczu – najczęściej 20 mikrogramów/80 mikrolitrów, dostępny w różnych objętościach wkładu lub wstrzykiwacza (np. 2,4 ml, 2,7 ml, 3 ml, 0,56 ml, 0,98 ml, 1,4 ml)891011121314
- Specjalne postacie do leczenia przewlekłej niedoczynności przytarczyc: wstrzykiwacze dostarczające indywidualnie dobierane dawki od 6 do 60 mikrogramów PTH(1-34) na dobę151617
Teryparatyd występuje jako jednoskładnikowy lek. Dostępne są także pochodne, takie jak palopegteryparatyd, stosowane w leczeniu niedoczynności przytarczyc15.
Najważniejsze wskazania
- Leczenie osteoporozy u kobiet po menopauzie z wysokim ryzykiem złamań
- Leczenie osteoporozy u mężczyzn z wysokim ryzykiem złamań
- Osteoporoza spowodowana długotrwałym przyjmowaniem glikokortykosteroidów
- Leczenie przewlekłej niedoczynności przytarczyc (palopegteryparatyd)181920
Dawkowanie – podstawowe informacje
Najczęściej stosowana dawka teryparatydu w leczeniu osteoporozy to 20 mikrogramów raz na dobę w postaci podskórnego wstrzyknięcia. Czas leczenia nie powinien przekraczać 24 miesięcy w ciągu życia pacjenta2122. W przypadku niedoczynności przytarczyc dawka jest ustalana indywidualnie i wymaga regularnej kontroli stężenia wapnia we krwi23.
Kiedy nie stosować teryparatydu?
- Nadwrażliwość na substancję czynną
- Ciąża i karmienie piersią
- Hiperkalcemia (zbyt wysokie stężenie wapnia we krwi)
- Ciężka niewydolność nerek
- Metaboliczne choroby kości (z wyjątkiem pierwotnej osteoporozy i osteoporozy polekowej)
- Stan po radioterapii kośćca
- Nowotwory układu kostno-szkieletowego lub przerzuty do kości
- Wzrost aktywności fosfatazy zasadowej o niewyjaśnionej przyczynie242526
Profil bezpieczeństwa
Teryparatyd nie powinien być stosowany przez kobiety w ciąży ani podczas karmienia piersią. U osób starszych zwykle nie jest wymagana modyfikacja dawki. Lek nie jest zalecany dla dzieci i młodzieży. U pacjentów z umiarkowaną niewydolnością nerek lub niewydolnością wątroby należy zachować ostrożność2728. Teryparatyd może powodować przemijające uczucie osłabienia lub zawroty głowy, dlatego należy zachować ostrożność podczas prowadzenia pojazdów i obsługiwania maszyn29.
Przedawkowanie – co warto wiedzieć?
Przedawkowanie teryparatydu może prowadzić do hiperkalcemii, nudności, wymiotów, zawrotów głowy i osłabienia. W przypadku podejrzenia przedawkowania należy odstawić lek i skontaktować się z lekarzem, a leczenie polega na kontrolowaniu poziomu wapnia i odpowiednim nawodnieniu303132.
Najważniejsze interakcje
- Ostrożność przy stosowaniu z glikozydami naparstnicy (np. digoksyna) – zwiększone ryzyko toksyczności przy wysokim poziomie wapnia
- Nie stwierdzono istotnych interakcji z hydrochlorotiazydem, raloksyfenem, hormonalną terapią zastępczą333435
Najczęstsze działania niepożądane
- Nudności
- Bóle kończyn
- Bóle i zawroty głowy
- Osłabienie lub zmęczenie
- Reakcje w miejscu wstrzyknięcia (zaczerwienienie, świąd, ból)
- Hiperkalcemia
- Rzadziej: reakcje alergiczne, niedociśnienie ortostatyczne, parestezje, ból stawów, wysypka363738
Mechanizm działania
Teryparatyd jest fragmentem ludzkiego parathormonu, który stymuluje komórki kościotwórcze do budowy nowej tkanki kostnej, a także zwiększa wchłanianie wapnia w jelitach i jego zwrotne wchłanianie w nerkach. Efektem jest odbudowa kości i poprawa ich wytrzymałości1239.
Stosowanie w ciąży
Teryparatyd nie powinien być stosowany przez kobiety w ciąży oraz karmiące piersią2425.
Stosowanie u dzieci
Nie ustalono bezpieczeństwa ani skuteczności teryparatydu u dzieci i młodzieży. Nie jest on zalecany do stosowania w tej grupie wiekowej404142.
Stosowanie u kierowców
Po zastosowaniu teryparatydu mogą wystąpić przemijające zawroty głowy lub uczucie osłabienia, dlatego należy zachować ostrożność podczas prowadzenia pojazdów i obsługiwania maszyn2928.
Teryparatyd – porównanie substancji czynnych
Teryparatyd, abaloparatyd i romosozumab to nowoczesne leki stosowane w leczeniu osteoporozy, różniące się wskazaniami, bezpieczeństwem i mechanizmem działania. Porównywane substancje czynne – czym się...
czytaj więcej ❯❯- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO FORSTEO 20 mikrogramów/80 mikrolitrów roztwór do wstrzykiwań we wstrzykiwaczu 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu*. Jeden wstrzykiwacz 2,4 ml zawiera 600 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). *Teryparatyd, rhPTH(1-34), wytwarzany metodą rekombinacji DNA przez E.coli , ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań. Bezbarwny, przezroczysty roztwór.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWskazania do stosowania
4.1 Wskazania do stosowania FORSTEO jest wskazany dla dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy spowodowanej długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn, o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecaną dawką produktu leczniczego FORSTEO jest 20 mikrogramów, podawane raz na dobę. Całkowity maksymalny czas leczenia produktem FORSTEO wynosi 24 miesiące (patrz punkt 4.4). Przez całe życie u pacjenta nie należy powtarzać 24 miesięcznego okresu leczenia produktem FORSTEO. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Po zakończeniu terapii produktem FORSTEO, pacjenci mogą stosować inne metody leczenia osteoporozy. Populacje szczególne Pacjenci z zaburzeniami czynności nerek Nie stosować produktu FORSTEO u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując produkt u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
Pacjenci z zaburzeniami czynności wątroby Nie ma danych dotyczących stosowania produktu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność stosując produkt FORSTEO. Dzieci, młodzież i młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności produktu FORSTEO u dzieci i młodzieży w wieku poniżej 18 lat. Produktu FORSTEO nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Pacjenci w podeszłym wieku Nie jest konieczna modyfikacja dawki produktu zależnie od wieku pacjenta (patrz punkt 5.2). Sposób podania Produkt FORSTEO należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci muszą być poinformowani o właściwym sposobie wykonywania wstrzyknięcia (patrz punkt 6.6). Informacje dla pacjentów dotyczące prawidłowego sposobu użycia wstrzykiwacza dostępne są także w Instrukcji użycia.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclPrzeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1 Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6) Wcześniej ujawniona hiperkalcemia Ciężka niewydolność nerek Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie Stan po radioterapii zewnętrznej lub wewnętrznej kośćca Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania produktu i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki produktu FORSTEO. Nie jest konieczne rutynowe monitorowanie wapnia podczas stosowania produktu.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
FORSTEO może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania FORSTEO u osób z czynną kamicą moczową. FORSTEO należy stosować ostrożnie u osób z czynną lub niedawno przebytą kamicą moczową, ze względu na ryzyko zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem produktu leczniczego FORSTEO obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu produktu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienia ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania produktu u pacjentów z umiarkowaną niewydolnością nerek. Stosowanie u młodych dorosłych Dane dotyczące stosowania produktu leczniczego u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym są ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania produktu FORSTEO. W przypadku zajścia w ciążę należy przerwać stosowanie FORSTEO. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Zawartość sodu Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, to znaczy produkt uznaje się za „wolny od sodu”.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki produktu leczniczego FORSTEO nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że produkt FORSTEO powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje produktu FORSTEO i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie produktu FORSTEO i raloksyfenu lub hormonalnej terapii zastępczej nie zmieniało wpływu FORSTEO na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania produktu FORSTEO, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania produktu FORSTEO. Ciąża Stosowanie produktu FORSTEO jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu FORSTEO jest przeciwwskazane podczas karmienia piersią. Nie wiadomo czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ produktu na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn FORSTEO nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów mechanicznych i obsługiwać urządzeń mechanicznych do czasu ustąpienia tych objawów.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działan, bóle i zawroty głowy. iami niepożądanymi występującymi podczas stosowania produktu FORSTEO są nudności, bóle kończyn Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8 % pacjentów otrzymujących produkt leczniczy FORSTEO i u 84,5 % pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W tabeli poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do 1/10), niezbyt często (≥1/1 000 do 1/100), rzadko (≥1/10 000 do 1/1 000), bardzo rzadko ( 1/10 000).
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Zaburzenia krwi i układu chłonnegoCzęsto: niedokrwistość Zaburzenia układu immunologicznegoRzadko: anafilaksja Zaburzenia metabolizmu i odżywianiaCzęsto: hipercholesterolemiaNiezbyt często: hiperkalcemia większa niż 2,76 mmol/l, nadmierne stężenie kwasu moczowego wsurowicy krwiRzadko: hiperkalcemia większa niż 3,25 mmol/l Zaburzenia psychiczneCzęsto: depresja Zaburzenia układu nerwowegoCzęsto: zawroty głowy, ból głowy, rwa kulszowa, omdlenie Zaburzenia ucha i błędnikaCzęsto: zawroty głowy (spowodowane zaburzeniami błędnika) Zaburzenia sercaCzęsto: kołatanie sercaNiezbyt często: tachykardia Zaburzenia naczynioweCzęsto: niedociśnienie Zaburzenia układu oddechowego, klatki piersiowej i śródpiersiaCzęsto: dusznośćNiezbyt często: rozedma płuc Zaburzenia żołądka i jelitCzęsto: nudności, wymioty, przepuklina rozworu przełykowego, choroba refluksowa przełykuNiezbyt często: guzy krwawnicze Zaburzenia skóry i tkanki podskórnejCzęsto: zwiększona potliwość Zaburzenia mięśniowo-szkieletowe i tkanki łącznejBardzo często: ból kończynCzęsto: kurcze mięśniNiezbyt często: ból mięśni, ból stawów, kurcze lub ból* mięśni pleców Zaburzenia nerek i dróg moczowychNiezbyt często: nietrzymanie moczu, nadmierne wydzielanie moczu, nagłe parcie na pęcherz, kamica nerkowaRzadko: niewydolność nerek lub zaburzenia czynności nerek Zaburzenia ogólne i stany w miejscu podaniaCzęsto: zmęczenie, ból w klatce piersiowej, osłabienie, łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień, miejscowe zasinienie, świąd i niewielkie krwawienie w miejscu wstrzyknięciaNiezbyt często: rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia.Rzadko: możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostre zaburzenia oddychania (duszność), obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowej, obrzęki (głównie obwodowe) Badania diagnostyczneNiezbyt często: zwiększenie masy ciała, szmery sercowe, zwiększenie aktywności fosfatazy zasadowej - CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
* Silne kurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1 % większą w porównaniu z placebo: zawroty głowy (spowodowane zaburzeniami błędnika), nudności, bóle kończyn, zawroty głowy, depresja, duszność. FORSTEO powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8 % pacjentów stosujących produkt leczniczy FORSTEO i 0,7 % osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. W dużym badaniu klinicznym, u 2,8 % kobiet otrzymujących produkt leczniczy FORSTEO wykryto przeciwciała reagujące krzyżowo z teryparatydem.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Przeciwciała zazwyczaj wykrywano po 12 miesiącach leczenia, a ich miano zmniejszało się po odstawieniu produktu. Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Produkt FORSTEO podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu: ujawniająca się po pewnym czasie hiperkalcemia, ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania produktu na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu: Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 g). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie lub ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na produkt FORSTEO. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05 AA02 Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. ProduktFORSTEO (rhPTH(1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki. Rezultat działania farmakodynamicznego FORSTEO wspomaga proces tworzenia się kości. Jest stosowany w leczeniu osteoporozy. Wpływ produktu FORSTEO na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podawanie FORSTEO raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI). Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np. mała gęstość mineralna kości [np.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90 % pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T-score= -2,6 SD). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 IU witaminy D na dobę. Wyniki stosowania produktu FORSTEO przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 1). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy. Tabela 1
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Częstość występowania złamań u kobiet w okresie pomenopauzalnym: Placebo (N = 544) (%) FORSTEO (N = 541) (%) Ryzyko względne(95% CI)w porównaniu z placebo Nowe złamania kręgów 14,3 5,0 b 0,35 (1)a (0,22; 0,55) Wielokrotne złamania 4,9 1,1 b 0,23 kręgów (2)a (0,09; 0,60) Złamania pozakręgowe spowodowane zwiększonąłamliwością c 5,5 2,6d 0,47(0,25; 0,87) Poważne złamania pozakręgowe spowodowane zwiększoną łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber imiednicy) 3,9 1,5d 0,38(0,17; 0,86) - CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących Forsteo, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p 0,001 w porównaniu z placebo c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. d p 0,025 w porównaniu z placebo Po średnio 19 miesiącach leczenia, odnotowano zwiększenie gęstości mineralnej tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9 % i 4 % w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu terapii produktem FORSTEO, 1262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania produktu FORSTEO.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania produktu FORSTEO odnotowano zmniejszenie o 41 % (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83 % stosowano wcześniej leczenie osteoporozy), były leczone produktem FORSTEO w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5 %, 2,6 % i 3,9 %. W okresie pomiędzy 18. a 24.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4 %, 1,2 % i 1,6 %. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy 4 z losowym doborem uczestników, wzięło udział 1360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej Forsteo zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9% pacjentek, a 18,8% podczas badania przyjmowało jednocześnie glikokortykoidy. Ukończyło 24-miesięczną obserwację 1013 (74,5%) pacjentek.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykoidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1433 IU/dobę (1400 IU/dobę), a w grupie przyjmującej ryzedronian 1191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano radiografię kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4%) u pacjentek leczonych Forsteo i 64/533 (12,0%) u pacjentek leczonych ryzedronianem, ryzyko względne (95% CI) = 0,44 (0,29-0,68), P <0,0001. Skumulowana częstość występowania łącznych złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8% w grupie pacjentek leczonych Forsteo i 9,8% w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95% CI) = 0,48 (0,32-0,74), P=0,0009.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą wwyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia gęstość mineralna tkanki kostnej kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35 % pacjentów miało złamania kręgów a 59 % złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 IU witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5 % i 1 % w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność produktu FORSTEO wykazano w pierwszej 18 miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg na dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 IU witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69 % pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie produktu FORSTEO spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2 %) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4 %) (p<0,001).
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Stosowanie FORSTEO spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6 %) w porównaniu z alendronianem (2,2 %) (p<0,01), jak również szyjki kości udowej (3,7 %) w porównaniu z alendronianem (2,1 %) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7 %, 0,9 % i 0,4 %. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących FORSTEO wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7 %) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej FORSTEO (1,7 %) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0 %) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5 %) leczonej FORSTEO (p=0,84).
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących FORSTEO w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2 % w porównaniu −1,9 %; p<0,001, dla kości biodra (3,8 % w porównaniu 0,9 %; p=0,005). Nie wykazano istotnego wpływu na częstość złamań kości.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania produktu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania produktu FORSTEO. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie produktu FORSTEO zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce produktu FORSTEO w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Produkt leczniczy nie wykazywał działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano FORSTEO we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera ), a co za tym idzie klirens PTH(1-84).
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
6. DANE FARMACEUTYCZNE: 6.1 Wykaz substancji pomocniczych Kwas octowy lodowaty Octan sodu (bezwodny) Mannitol, Metakrezol Kwas solny (do ustalenia pH) Wodorotlenek sodu (do ustalenia pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 2 lata Wykazano trwałość chemiczną, fizyczną i mikrobiologiczną stosowanego produktu w okresie 28 dni w temperaturze 2-8ºC. Po otwarciu produkt leczniczy można przechowywać nie dłużej niż 28 dni w temperaturze 2ºC do 8ºC. Za inne warunki i czas przechowywania stosowanego produktu odpowiedzialność ponosi użytkownik. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2ºC – 8ºC). Bezpośrednio po każdym użyciu wstrzykiwacz należy ponownie umieścić w lodówce. Nie zamrażać. Nie przechowywać wstrzykiwacza z zamocowaną igłą.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
6.5 Rodzaj i zawartość opakowania Roztwór 2,4 ml we wkładzie (szkło silikonowane typu I) zamkniętym korkiem (z gumy halobutylowej), zatyczką (poliizopren/ laminat z gumy bromobutylowej/ aluminium) umieszczony w jednorazowym wstrzykiwaczu. FORSTEO jest dostępny w opakowaniach zawierających 1 lub 3 wstrzykiwacze. Jeden wstrzykiwacz zawiera 28 dawek po 20 mikrogramów każda (w 80 mikrolitrach). Nie wszystkie wielkości opakowań muszą się znajdować w obrocie. 6.6 Specjalne środki ostrożności dotyczące usuwania FORSTEO jest to jednorazowy wstrzykiwacz. Przeznaczony jest do stosowania przez jednego pacjenta. Do każdego wstrzyknięcia musi być użyta nowa jałowa igła. Do każdego opakowania dołączono Instrukcję użycia zawierającą dokładny opis sposobu użycia wstrzykiwacza. Do wstrzykiwaczy nie dołączono igieł. Wstrzykiwacz można stosować z igłami przeznaczonymi do wstrzykiwaczy z insuliną. Po każdym wstrzyknięciu, wstrzykiwacz należy ponownie umieścić w lodówce.
- CHPL leku Forsteo, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
Nie należy stosować produktu FORSTEO, jeżeli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Należy także zapoznać się z informacjami o sposobie użycia wstrzykiwacza, zamieszczonymi w instrukcji użycia. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Terrosa 20 mikrogramów/80 mikrolitrów, roztwór do wstrzykiwań 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu*. Jeden wkład z 2,4 ml roztworu zawiera 600 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). *Teryparatyd, rhPTH(1-34), wytwarzany metodą rekombinacji DNA przez E.coli , ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (iniekcji). Bezbarwny, przezroczysty roztwór do wstrzykiwań o pH 3,8 – 4,5.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Terrosa jest wskazany dla pacjentów dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy związanej z długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn, o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecana dawka produktu leczniczego Terrosa to 20 mikrogramów, podawane jeden raz na dobę. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując produkty zawierające wapń i witaminę D. Całkowity maksymalny czas leczenia teryparatydem wynosi 24 miesiące (patrz punkt 4.4). Przez całe życie u pacjenta nie należy powtarzać 24 miesięcznego okresu leczenia teryparatydem. Po zakończeniu leczenia teryparatydem, pacjenci mogą stosować inne metody leczenia osteoporozy. Populacje szczególne Zaburzenia czynności nerek Nie wolno stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując teryparatyd u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDawkowanie
Zaburzenia czynności wątroby Nie ma danych dotyczących stosowania teryparatydu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność podczas stosowania teryparatydu. Dzieci i młodzież oraz młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Teryparatydu nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Pacjenci w podeszłym wieku Nie jest konieczna modyfikacja dawki produktu zależnie od wieku pacjenta (patrz punkt 5.2). Sposób podawania Produkt leczniczy Terrosa należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci powinni zostać przeszkoleni w zakresie właściwego sposobu wykonywania wstrzyknięcia.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDawkowanie
W celu zapoznania się z instrukcjami dotyczącymi produktu leczniczego przed podaniem patrz punkt 6.6 oraz instrukcja użytkowania na końcu ulotki. Instrukcje użytkowania wstrzykiwacza Terrosa dołączone do wstrzykiwacza także mogą być stosowane do poinstruowania pacjentów jak prawidłowo stosować wstrzykiwacz.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1. - Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6). - Wcześniej ujawniona hiperkalcemia. - Ciężkie zaburzenia czynności nerek. - Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów. - Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie. - Stan po radioterapii zewnętrznej lub wewnętrznej kośćca. - Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki teryparatydu. Nie jest konieczne rutynowe monitorowanie wapnia podczas leczenia.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclSpecjalne środki ostrozności
Teryparatyd może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u pacjentów z czynną kamicą moczową. Teryparatyd należy stosować ostrożnie u pacjentów z czynną lub niedawno przebytą kamicą moczową, ze względu na możliwość zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienie ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclSpecjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Stosowanie u młodych dorosłych Doświadczenie związane ze stosowaniem u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym, jest ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania teryparatydu. W przypadku zajścia w ciążę należy przerwać stosowanie teryparatydu. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclSpecjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Substancja pomocnicza Lek zawiera mniej niż 1 mmol sodu (23 mg) w jednej dawce, to znaczy lek uznaje się za „wolny od sodu”.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie raloksyfenu lub hormonalnej terapii zastępczej z teryparatydem nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania teryparatydu, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania teryparatydu. Ciąża Stosowanie teryparatydu jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu leczniczego Terrosa jest przeciwwskazane podczas karmienia piersią. Nie wiadomo, czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Teryparatyd nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów i obsługiwać maszyn do czasu ustąpienia tych objawów.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi występującymi podczas stosowania teryparatydu są nudności, bóle kończyn, bóle i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8% pacjentów otrzymujących teryparatyd i u 84,5% pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W Tabeli 1 podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100) oraz rzadko (≥1/10 000 do <1/1 000). Tabela 1. Działania niepożądane
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDziałania niepożądane
Klasyfikacjaukładów i narządów Bardzo często Często Niezbyt często Rzadko Zaburzenia krwi iukładu chłonnego Niedokrwistość Zaburzeniaukładuimmunologiczneg o Anafilaksja Zaburzenia metabolizmu i odżywiania Hipercholesterole mia Hiperkalcemia większa niż 2,76 mmol/lhiperurykemia Hiperkalcemia większa niż 3,25 mmol/l Zaburzeniapsychiczne Depresja Zaburzenia układu nerwowego Zawroty głowy, ból głowy, rwa kulszowa,omdlenie Zaburzenia ucha ibłędnika Zawroty głowypochodzeniabłędnikowego Zaburzenia serca Kołatanie serca Tachykardia Zaburzenia naczyniowe Niedociśnienie tętnicze Zaburzenia układu oddechowego, klatki piersiowej iśródpiersia Duszność Rozedma płuc Zaburzeniażołądka i jelit Nudności, wymioty, przepuklina rozworu przełykowego, choroba refleksowaprzełyku Guzki krwawnicze Zaburzenia skóry i tkanki podskórnej Zwiększona potliwość Zaburzenia mięśniowo- szkieletowe itkanki łącznej Ból kończyn Skurcze mięśni Ból mięśni, bólstawów, skurczelub ból* mięśni pleców Zaburzenia nerek i dróg moczowych Nietrzymanie moczy, nadmierne wydzielaniemoczu, nagłe Niewydolność/zab urzenia czynności nerek - CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDziałania niepożądane
Klasyfikacja układów i narządów Bardzo często Często Niezbyt często Rzadko parcie na pęcherz,kamica nerkowa Zaburzenia ogólne i stany w miejscu podania Zmęczenie, ból w klatce piersiowej, osłabienie, łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień, miejscowe zasinienie, świąd i niewielkie krwawienie w miejscuwstrzyknięcia Rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia. Możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostra duszność, obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowej, obrzęki (głównie obwodowe) Badania diagnostyczne Zwiększenie masy ciała, szmery sercowe, zwiększenie aktywności fosfatazyzasadowej - CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDziałania niepożądane
* Silne skurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1% większą w porównaniu z placebo: zawroty głowy pochodzenia błędnikowego, nudności, bóle kończyn, zawroty głowy pochodzenia ośrodkowego, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8 % pacjentów stosujących teryparatyd i 0,7 % osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. W dużym badaniu klinicznym, u 2,8 % kobiet otrzymujących teryparatyd wykryto przeciwciała reagujące krzyżowo z teryparatydem.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclDziałania niepożądane
Przeciwciała zazwyczaj wykrywano po 12 miesiącach leczenia, a ich miano zmniejszało się po odstawieniu produktu. Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu obejmują ujawniającą się po pewnym czasie hiperkalcemię oraz ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 mikrogramów). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie i (lub) ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na teryparatyd. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05AA02 Terrosa jest produktem leczniczym biopodobnym. Szczegółowe informacje są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu. Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rhPTH(1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Rezultat działania farmakodynamicznego Teryparatyd wspomaga proces tworzenia się kości i jest stosowany w leczeniu osteoporozy. Wpływ teryparatydu na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy. Podawanie teryparatydu raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI).
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np. mała gęstość mineralna kości [np. wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90 % pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T-score= -2,6 SD). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 j.m.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 2). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy. Tabela 2. Częstość występowania złamań u kobiet w wieku pomenopauzalnym
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Placebo (N = 544) (%) Teryparatyd (N = 541) (%) Ryzyko względna(95% CI) wporównaniu do placebo Nowe złamania kręgów (≥1) a 14,3 5,0 b 0,35(0,22, 0,55) Wielokrotne złamaniakręgów (≥2) a 4,9 1,1 b 0,23(0,09, 0,60) Złamania pozakręgowe spowodowane zwiększonąłamliwością c 5,5% 2,6% d 0,47(0,25, 0,87) Ciężkie złamania pozakręgowe spowodowane zwiększoną łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber imiednicy) 3,9% 1,5% d 0,38(0,17, 0,86) - CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności. a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących teryparatyd, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p≤0,001 w porównaniu z placebo. c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. d p≤0,025 w porównaniu z placebo. Po średnio 19 miesiącach leczenia, odnotowano zwiększenie BMD lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9 % i 4 % w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu leczenia teryparatydem, 1 262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41% (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83 % stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5 %, 2,6 % i 3,9 %. W okresie pomiędzy 18. a 24.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4 %, 1,2 % i 1,6 %. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy IV z losowym doborem uczestników, wzięło udział 1 360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9% pacjentek, a 18,8% podczas badania przyjmowało jednocześnie glikokortyksteroidy. 24-miesięczną obserwację ukończyło 1013 (74,5%) pacjentek.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykosteroidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1433 IU/dobę (1400 IU/dobę), a w grupie przyjmującej ryzedronian 1191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano badanie rentgenowskie kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4%) u pacjentek leczonych teryparatydemi i 64/533 (12,0%) u pacjentek leczonych ryzedronianem, ryzyko względne (95%CI) = 0,44 (0,29-0,68), p<0,0001. Łączna, skumulowana częstość występowania złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8% w grupie pacjentek leczonych teryparatydemi 9,8% w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95% CI) = 0,48 (0,32-0,74), p=0,0009.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą w wyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia BMD kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35% pacjentów miało złamania kręgów a 59 % złamania pozakręgowe. Wszystkim uczestnikom podawano 1 000 mg wapnia na dobę oraz co najmniej 400 j.m. witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5 % i 1 % w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18-miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg/dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1 000 mg wapnia na dobę i 800 j.m. witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24% pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69% pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2 %) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4 %) (p<0,001).
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6 %) w porównaniu z alendronianem (2,2 %) (p<0,01), jak również szyjki kości udowej (3,7 %) w porównaniu z alendronianem (2,1 %) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7 %, 0,9 % i 0,4 %. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7 %) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7 %) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0 %) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5 %) leczonej teryparatydem (p=0,84).
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2 % w porównaniu −1,9 %; p<0,001, dla kości biodra (3,8 % w porównaniu 0,9 %; p=0,005). Nie wykazano jednak istotnego wpływu na częstość złamań kości.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania teryparatydu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Nie wykazywał także działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1 000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Terrosa, roztwór do wstrzykiwań, 20 mcg/ 80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Movymia 20 mikrogramów/80 mikrolitrów, roztwór do wstrzykiwań 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu*. Jeden wkład z 2,4 ml roztworu zawiera 600 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). *Teryparatyd, rhPTH(1-34), wytwarzany metodą rekombinacji DNA przez E.coli , ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (iniekcji). Bezbarwny, przezroczysty roztwór do wstrzykiwań o pH 3,8 – 4,5.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Movymia jest wskazany dla pacjentów dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy związanej z długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn, o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecana dawka produktu leczniczego Movymia to 20 mikrogramów, podawane jeden raz na dobę. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Całkowity maksymalny czas leczenia teryparatydem wynosi 24 miesiące (patrz punkt 4.4). Przez całe życie u pacjenta nie należy powtarzać 24 miesięcznego okresu leczenia teryparatydem. Po zakończeniu leczenia teryparatydem, pacjenci mogą stosować inne metody leczenia osteoporozy. Populacje szczególne Zaburzenia czynności nerek Nie wolno stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując teryparatyd u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
Zaburzenia czynności wątroby Nie ma danych dotyczących stosowania teryparatydu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność podczas stosowania teryparatydu. Dzieci i młodzież oraz młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Teryparatydu nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Pacjenci w podeszłym wieku Nie jest konieczna modyfikacja dawki produktu zależnie od wieku pacjenta (patrz punkt 5.2). Sposób podawania Produkt leczniczy Movymia należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci powinni zostać przeszkoleni w zakresie właściwego sposobu wykonywania wstrzyknięcia.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
W celu zapoznania się z instrukcjami dotyczącymi produktu leczniczego przed podaniem patrz punkt 6.6 oraz instrukcja użytkowania na końcu ulotki. Instrukcje użytkowania wstrzykiwacza Movymia dołączone do wstrzykiwacza także mogą być stosowane do poinstruowania pacjentów jak prawidłowo stosować wstrzykiwacz.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclPrzeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1. Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6). Wcześniej ujawniona hiperkalcemia. Ciężkie zaburzenia czynności nerek. Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów. Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie. Stan po radioterapii zewnętrznej lub wewnętrznej kośćca. Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki teryparatydu. Nie jest konieczne rutynowe monitorowanie wapnia podczas leczenia.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
Teryparatyd może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u pacjentów z czynną kamicą moczową. Teryparatyd należy stosować ostrożnie u pacjentów z czynną lub niedawno przebytą kamicą moczową, ze względu na możliwość zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienie ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Stosowanie u młodych dorosłych Doświadczenie związane ze stosowaniem u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym, jest ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania teryparatydu. W przypadku zajścia w ciążę należy przerwać stosowanie teryparatydu. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Substancja pomocnicza (sodu) Lek zawiera mniej niż 1 mmol (23 mg) sodu na jednostka dawkowania, to znaczy lek uznaje się za „wolny od sodu”.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie raloksyfenu lub hormonalnej terapii zastępczej z teryparatydem nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania teryparatydu, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania teryparatydu. Ciąża Stosowanie teryparatydu jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu leczniczego Movymia jest przeciwwskazane podczas karmienia piersią. Nie wiadomo, czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Teryparatyd nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów i obsługiwać maszyn do czasu ustąpienia tych objawów.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi występującymi podczas stosowania teryparatydu są nudności, bóle kończyn, bóle i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8 % pacjentów otrzymujących teryparatyd i u 84,5 % pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W Tabeli 1 poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100) oraz rzadko (≥1/10 000 do <1/1 000). Tabela 1. Działania niepożądane
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Klasyfikacjaukładów i narządów Bardzoczęsto Często Niezbyt często Rzadko Zaburzenia krwi iukładu chłonnego Niedokrwistość Zaburzenia układuimmunologicznego Anafilaksja Zaburzenia metabolizmu i odżywiania Hipercholesterolemia Hiperkalcemia większa niż 2,76 mmol/lhiperurykemia Hiperkalcemia większa niż 3,25 mmol/l Zaburzenia psychiczne Depresja Zaburzenia układunerwowego Zawroty głowy, ból głowy, rwakulszowa, omdlenie Zaburzenia ucha ibłędnika Zawroty głowypochodzeniabłędnikowego Zaburzenia serca Kołatanie serca Tachykardia Zaburzenia naczyniowe Niedociśnienie tętnicze Zaburzenia układu oddechowego, klatki piersiowej iśródpiersia Duszność Rozedma płuc Zaburzenia żołądkai jelit Nudności, wymioty, przepuklina rozworu przełykowego, choroba refleksowaprzełyku Guzy krwawnicze Zaburzenia skóry i tkanki podskórnej Zwiększona potliwość Zaburzenia mięśniowo- szkieletowe i tkankiłącznej Bólkończyn Skurcze mięśni Ból mięśni, ból stawów, skurcze lub ból* mięśnipleców Zaburzenia nerek i dróg moczowych Nietrzymanie moczy, nadmierne wydzielanie moczu, nagłe parcie na pęcherz,kamica nerkowa Niewydolność/zab urzenia czynności nerek Zaburzenia ogólne i stany w miejscu podania Zmęczenie, ból w klatce piersiowej, osłabienie, łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień, miejscowe zasinienie, świąd i niewielkie krwawienie w miejscuwstrzyknięcia Rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia. Możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostra duszność, obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowej, obrzęki (głównieobwodowe) - CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Klasyfikacjaukładów i narządów Bardzoczęsto Często Niezbyt często Rzadko Badania diagnostyczne Zwiększenie masy ciała, szmery sercowe, zwiększenie aktywności fosfatazyzasadowej - CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
* Silne skurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1 % większą w porównaniu z placebo: zawroty głowy pochodzenia błędnikowego, nudności, bóle kończyn, zawroty głowy pochodzenia ośrodkowego, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8 % pacjentów stosujących teryparatyd i 0,7 % osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. W dużym badaniu klinicznym, u 2,8 % kobiet otrzymujących teryparatyd wykryto przeciwciała reagujące krzyżowo z teryparatydem.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Przeciwciała zazwyczaj wykrywano po 12 miesiącach leczenia, a ich miano zmniejszało się po odstawieniu produktu. Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu obejmują ujawniającą się po pewnym czasie hiperkalcemię oraz ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 mikrogramów). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie i (lub) ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na teryparatyd. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05AA02 Movymia jest produktem leczniczym biopodobnym. Szczegółowe informacje są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu . Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rhPTH(1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Rezultat działania farmakodynamicznego Teryparatyd wspomaga proces tworzenia się kości i jest stosowany w leczeniu osteoporozy. Wpływ teryparatydu na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy. Podawanie teryparatydu raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI).
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np. mała gęstość mineralna kości [np. wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90 % pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T-score= -2,6 SD). Wszystkim pacjentkom podawano 1 000 mg wapnia na dobę i przynajmniej 400 j.m.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 2). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy. Tabela 2. Częstość występowania złamań u kobiet w wieku pomenopauzalnym
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Placebo (N = 544) (%) Teryparatyd (N = 541) (%) Ryzyko względna(95% CI) wporównaniu do placebo Nowe złamania kręgów (≥1) a 14,3 5,0 b 0,35(0,22, 0,55) Wielokrotne złamania kręgów (≥2) a 4,9 1,1 b 0,23(0,09, 0,60) Złamania pozakręgowe spowodowane zwiększoną łamliwością c 5,5% 2,6% d 0,47(0,25, 0,87) Ciężkie złamania pozakręgowe spowodowane zwiększoną łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber imiednicy) 3,9% 1,5% d 0,38(0,17, 0,86) - CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności. a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących teryparatyd, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p≤0,001 w porównaniu z placebo. c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. d p≤0,025 w porównaniu z placebo. Po średnio 19 miesiącach leczenia, odnotowano zwiększenie BMD lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9 % i 4 % w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu leczenia teryparatydem, 1 262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41 % (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83 % stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5 %, 2,6 % i 3,9 %. W okresie pomiędzy 18. a 24.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4 %, 1,2 % i 1,6 %. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy IV z losowym doborem uczestników, wzięło udział 1 360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9 % pacjentek, a 18,8 % podczas badania przyjmowało jednocześnie glikokortykosteroidy. 24-miesięczną obserwację ukończyło 1 013 (74,5 %) pacjentek.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykosteroidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1 433 IU/dobę (1 400 IU/dobę), a w grupie przyjmującej ryzedronian 1 191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano badanie rentgenowskie kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4 %) u pacjentek leczonych teryparatydem i 64/533 (12,0 %) u pacjentek leczonych ryzedronianem, ryzyko względne (95 % CI) = 0,44 (0,29 - 0,68), p<0,0001. Łączna, skumulowana częstość występowania złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8 % w grupie pacjentek leczonych teryparatydem i 9,8 % w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95 % CI) = 0,48 (0,32- 0,74), p=0,0009.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą w wyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia BMD kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35 % pacjentów miało złamania kręgów a 59 % złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 j.m. witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5 % i 1 % w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18-miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg/dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 j.m. witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69 % pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2 %) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4 %) (p<0,001).
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6 %) w porównaniu z alendronianem (2,2 %) (p<0,01), jak również szyjki kości udowej (3,7 %) w porównaniu z alendronianem (2,1 %) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7 %, 0,9 % i 0,4 %. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7 %) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7 %) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0 %) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5 %) leczonej teryparatydem (p=0,84).
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2 % w porównaniu −1,9 %; p<0,001, dla kości biodra (3,8 % w porównaniu 0,9 %; p=0,005). Nie wykazano jednak istotnego wpływu na częstość złamań kości.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania teryparatydu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Nie wykazywał także działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1 000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Movymia, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Osteoteri, 20 mikrogramów/80 mikrolitrów, roztwór do wstrzykiwań we wstrzykiwaczu. 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Każda dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu. Teryparatyd, wytwarzany syntetycznie, ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Jeden wkład z 2,4 ml roztworu zawiera 600 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań. Bezbarwny, przezroczysty, izotoniczny roztwór o pH 3,8 – 4,5.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Osteoteri jest wskazany dla pacjentów dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy związanej z długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecana dawka produktu leczniczego Osteoteri to 20 mikrogramów, podawane jeden raz na dobę. Całkowity maksymalny czas leczenia teryparatydem wynosi 24 miesiące (patrz punkt 4.4). Nie należy powtarzać 24-miesięcznego okresu leczenia teryparatydem przez całe życie pacjenta. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Po zakończeniu leczenia teryparatydem, u pacjentów można stosować inne metody leczenia osteoporozy. Populacje szczególne Zaburzenia czynności nerek. Nie wolno stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując teryparatyd u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek. Zaburzenia czynności wątroby.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDawkowanie
Nie ma danych dotyczących stosowania teryparatydu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność podczas stosowania teryparatydu. Dzieci i młodzież oraz młodzi dorośli przed zakończeniem rozwoju nasad kości długich. Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Teryparatydu nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych przed zakończeniem rozwoju nasad kości długich. Pacjenci w podeszłym wieku. Nie jest konieczna modyfikacja dawki zależnie od wieku pacjenta (patrz punkt 5.2). Sposób podawania Produkt leczniczy Osteoteri należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Przed pierwszym użyciem wstrzykiwacza należy uwolnić dawkę wstępną. Pacjenci muszą być poinformowani o właściwym sposobie wykonywania wstrzyknięcia (patrz punkt 6.6).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDawkowanie
Informacje dla pacjentów dotyczące prawidłowego sposobu użycia wstrzykiwacza dostępne są także w instrukcji obsługi.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1. Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6). Wcześniej ujawniona hiperkalcemia. Ciężkie zaburzenia czynności nerek. Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów. Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie. Stan po radioterapii zewnętrznej lub wewnętrznej kośćca. Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki teryparatydu. Nie jest konieczne rutynowe monitorowanie wapnia podczas leczenia. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u pacjentów z czynną kamicą moczową. Teryparatyd należy stosować ostrożnie u pacjentów z czynną lub niedawno przebytą kamicą moczową, ze względu na możliwość zaostrzenia przebiegu tej choroby.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienia ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu. Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Stosowanie u młodych dorosłych Doświadczenie związane ze stosowaniem u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym, jest ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania teryparatydu. W przypadku zajścia w ciążę należy przerwać stosowanie teryparatydu. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj. 24 miesięcy, do czasu uzyskania nowych danych klinicznych. Produkt leczniczy Osteoteri zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że zasadniczo nie zawiera sodu.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie raloksyfenu lub hormonalnej terapii zastępczej z teryparatydem nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania teryparatydu, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania teryparatydu. Ciąża Stosowanie teryparatydu jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersi? Stosowanie produktu leczniczego Osteoteri jest przeciwwskazane podczas karmienia piersią. Nie wiadomo, czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Teryparatyd nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów i obsługiwać maszyn do czasu ustąpienia tych objawów.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi występującymi podczas stosowania teryparatydu są nudności, bóle kończyn, bóle i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8% pacjentów otrzymujących teryparatyd i u 84,5% pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W tabeli poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1 000), bardzo rzadko (<1/10 000).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Zaburzenia krwi i układu chłonnego Często: niedokrwistość Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia Często: duszność Niezbyt często: rozedma płuc Zaburzenia układu immunologicznego Rzadko: anafilaksja Zaburzenia żołądka i jelit Często: nudności, wymioty, przepuklina rozworu przełykowego, choroba refluksowa przełyku Niezbyt często: guzy krwawnicze Zaburzenia metabolizmu i odżywiania Często: hipercholesterolemia Niezbyt często: hiperkalcemia większa niż 2,76 mmol/l, nadmierne stężenie kwasu moczowego w surowicy krwi Rzadko: hiperkalcemia większa niż 3,25 mmol/l Zaburzenia skóry i tkanki podskórnej Często: zwiększona potliwość Zaburzenia psychiczne Często: depresja Zaburzenia mięśniowo-szkieletowe i tkanki łącznej Bardzo często: ból kończyn Często: kurcze mięśni Niezbyt często: ból mięśni, ból stawów, kurcze lub ból mięśni pleców Zaburzenia układu nerwowego Często: zawroty głowy, ból głowy, rwa kulszowa, omdlenie Zaburzenia nerek i dróg moczowych Niezbyt często: nietrzymanie moczu, nadmierne wydzielanie moczu, nagłe parcie na pęcherz, kamica nerkowa Rzadko: niewydolność nerek lub zaburzenia czynności nerek Zaburzenia ucha i błędnika Często: zawroty głowy Zaburzenia ogólne i stany w miejscu podania Często: zmęczenie, ból w klatce piersiowej, osłabienie, łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień, miejscowe zasinienie, świąd i niewielkie krwawienie w miejscu wstrzyknięcia Niezbyt często: rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Rzadko: możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostre zaburzenia oddychania (duszność), obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowej, obrzęki (głównie obwodowe) Zaburzenia serca Często: kołatanie serca Niezbyt często: tachykardia Badania diagnostyczne Niezbyt często: zwiększenie masy ciała, szmery sercowe, zwiększenie aktywności fosfatazy zasadowej Zaburzenia naczyniowe Często: niedociśnienie * Silne skurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością o ≥ 1% większą w porównaniu z placebo: zawroty głowy pochodzenia błędnikowego, nudności, bóle kończyn, zawroty głowy pochodzenia ośrodkowego, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Podczas badań klinicznych u 2,8% pacjentów stosujących teryparatyd i 0,7% osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. W dużym badaniu klinicznym, u 2,8% kobiet otrzymujących teryparatyd wykryto przeciwciała reagujące krzyżowo z teryparatydem. Przeciwciała zazwyczaj wykrywano po 12 miesiącach leczenia, a ich miano zmniejszało się po odstawieniu produktu. Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301 faks: + 48 22 49 21 309 Strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu, obejmują ujawniającą się po pewnym czasie hiperkalcemię oraz ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania na podstawie spontanicznych zgłoszeń po wprowadzeniu do obrotu Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 mikrogramów). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie i (lub) ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania nie obserwowano żadnych działań niepożądanych.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na teryparatyd. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05 AA02 Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rhPTH(1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki. Działania farmakodynamiczne Teryparatyd wspomaga proces tworzenia się kości stosowany w leczeniu osteoporozy. Wpływ teryparatydu na układ kostny zależy od przebiegu ekspozycji ogólnoustrojowej.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podawanie teryparatydu raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka, takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI). Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np. mała gęstość mineralna kości [np.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90% pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm2 (co odpowiadało wartości wskaźnika T-score= -2,6). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 IU witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszenie częstości złamań (Tabela 1). Aby zapobiec jednemu lub więcej nowym złamaniom kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Tabela 1 Częstość występowania złamań u kobiet w wieku pomenopauzalnym Placebo (N = 544) (%) Teryparatyd (N = 541) (%) Ryzyko względne (95% CI) w porównaniu do placebo Nowe złamania kręgów (≥1) 14,3 5,0 0,35 (0,22, 0,55) Wielokrotne złamania kręgów (≥2) 4,9 1,1 0,23 (0,09, 0,60) Złamania pozakręgowe spowodowane zwiększoną łamliwości? 5,5% 2,6% 0,47 (0,25, 0,87) Ciężkie złamania pozakręgowe spowodowane zwiększoną łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber i miednicy) 3,9% 1,5% 0,38 (0,17, 0,86) Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności. Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących teryparatyd, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. p≤0,001 w porównaniu z placebo.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. p≤0,025 w porównaniu z placebo. Po średnio 19 miesiącach leczenia, odnotowano zwiększenie gęstości mineralnej tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9% i 4% w porównaniu z placebo (p<0,001). Postępowanie po leczeniu: Po zakończeniu leczenia teryparatydem, 1262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu. Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41% (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83% stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5%, 2,6% i 3,9%. W okresie pomiędzy 18. a 24. miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4%, 1,2% i 1,6%. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy 4 z losowym doborem uczestników, wzięło udział 1360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9% pacjentek, a 18,8% podczas badania przyjmowało jednocześnie glikokortykoidy. Ukończyło 24-miesięczną obserwację 1013 (74,5%) pacjentek. Średnia (mediana) skumulowana dawka glukokortyidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1433 IU/dobę (1400 IU/dobę), a w grupie przyjmującej ryzedronian 1191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano radiografię kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4%) u pacjentek leczonych teryparatydem i 64/533 (12,0%) u pacjentek leczonych ryzedronianem, ryzyko względne (95% CI) = 0,44 (0,29-0,68), p<0,0001.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Skumulowana częstość występowania łącznych złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8% w grupie pacjentek leczonych teryparatydem i 9,8% w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95% CI) = 0,48 (0,32-0,74), p=0,0009. Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą w wyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia gęstość mineralna tkanki kostnej kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35% pacjentów miało złamania kręgów a 59% złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 IU witaminy D na dobę.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5% i 1% w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań. Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18-miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem leku porównawczego (alendronian w dawce 10 mg/dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28% pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 IU witaminy D na dobę.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przedmenopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. W punkcie wyjściowym średni wiek kobiet w okresie przedmenopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24% pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69% pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2%) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4%) (p<0,001). Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6%) w porównaniu z alendronianem (2,2%) (p<0,01), jak również szyjki kości udowej (3,7%) w porównaniu z alendronianem (2,1%) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7%, 0,9% i 0,4%. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7%) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7%) (p=0,01).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0%) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5%) leczonej teryparatydem (p=0,84). U kobiet w okresie przedmenopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2% w porównaniu −1,9%; p<0,001, dla kości biodra (3,8% w porównaniu 0,9%; p=0,005). Nie wykazano jednak istotnego wpływu na częstość złamań kości.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania teryparatydu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi wchłaniania z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Nie wykazywał także działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1000 µg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 µg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Kwas octowy lodowaty (E 260) Sodu octan bezwodny (E 262) Mannitol (E 421) Metakrezol Kwas solny (E 507) (do ustalenia pH) Sodu wodorotlenek (E 524) (do ustalenia pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 2 lata Wykazano trwałość chemiczną, fizyczną i mikrobiologiczną w okresie 28 dni w temperaturze 2-8ºC. Po otwarciu produkt leczniczy może być przechowywany nie dłużej niż 28 dni w temperaturze 2ºC do 8ºC. Za inne warunki i czas przechowywania stosowanego produktu odpowiedzialność ponosi użytkownik. 6.4 Specjalne środki ostrożności podczas przechowywania Wstrzykiwacz należy zamknąć nasadką po użyciu (ze względu na wrażliwość roztworu do wstrzykiwań na światło). Przechowywać w lodówce (2ºC – 8ºC).
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
Bezpośrednio po każdym użyciu wstrzykiwacz należy ponownie umieścić w lodówce. Nie zamrażać. Nie przechowywać wstrzykiwacza z założoną igłą. 6.5 Rodzaj i zawartość opakowania Wkład zawierający 2,4 ml roztworu (szkło silikonowane typu I) z tłokiem, zamknięty gumowym korkiem i aluminiowym wieczkiem, umieszczony we wstrzykiwaczu. Produkt leczniczy Osteoteri jest dostępny w opakowaniach zawierających 1 lub 3 wkłady. Każdy wkład zawiera 28 dawek po 20 mikrogramów każda (w 80 mikrolitrach). Nie wszystkie wielkości opakowań muszą się znajdować w obrocie. 6.6 Specjalne środki ostrożności dotyczące usuwania Produkt leczniczy Osteoteri to wstrzykiwacz. Przeznaczony jest do stosowania przez jednego pacjenta. Do każdego wstrzyknięcia musi być użyta nowa jałowa igła. Do każdego opakowania dołączono instrukcję obsługi zawierającą dokładny opis sposobu użycia wstrzykiwacza. Do wstrzykiwaczy nie dołączono igieł. Wstrzykiwacz można stosować z igłami przeznaczonymi do wstrzykiwaczy z insuliną.
- CHPL leku Osteoteri, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
Po każdym wstrzyknięciu, wstrzykiwacz należy ponownie umieścić w lodówce. Nie należy stosować produktu Osteoteri, jeżeli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Należy także zapoznać się z informacjami o sposobie użycia wstrzykiwacza, zamieszczonymi w instrukcji obsługi. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Nazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Livogiva 20 mikrogramów/80 mikrolitrów roztwór do wstrzykiwań we wstrzykiwaczu półautomatycznym napełnionym 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu*. Każdy wstrzykiwacz półautomatyczny napełniony 2,7 mL zawiera 675 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). *Teryparatyd, rhPTH(1-34), wytwarzany metodą rekombinacji DNA przez P. fluorescens , ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań. Bezbarwny, przezroczysty roztwór.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Wskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Livogiva jest wskazany dla dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy spowodowanej długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Dawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecaną dawką produktu leczniczego Livogiva jest 20 mikrogramów, podawane raz na dobę. Całkowity maksymalny czas leczenia produktem Livogiva wynosi 24 miesiące (patrz punkt 4.4). 24-miesięcznego okresu leczenia produktem Livogiva nie należy powtarzać przez całe życie pacjenta. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Po zakończeniu terapii produktem Livogiva, pacjenci mogą stosować inne metody leczenia osteoporozy. Populacje szczególne Pacjenci w podeszłym wieku Nie jest konieczna modyfikacja dawki produktu zależnie od wieku pacjenta (patrz punkt 5.2). Zaburzenia czynności nerek Nie stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując teryparatyd u pacjentów z umiarkowanymi zaburzeniami czynności nerek (patrz punkt 4.4).
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Dawkowanie
Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek. Zaburzenia czynności wątroby Nie ma danych dotyczących pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność stosując teryparatyd. Dzieci, młodzież i młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Teryparatydu nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Sposób podania Produkt Livogiva należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci muszą zostać przeszkoleni odnośnie właściwego sposobu wykonywania wstrzyknięcia (patrz punkt 6.6). Należy także zapoznać się z informacjami dotyczącymi prawidłowego sposobu użycia wstrzykiwacza, zamieszczonymi w instrukcji użycia.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Przeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1 Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6) Wcześniej ujawniona hiperkalcemia Ciężka niewydolność nerek Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie Stan po radioterapii zewnętrznej lub wewnętrznej kośćca Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Specjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania produktu i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki produktu Livogiva. Nie jest konieczne rutynowe monitorowanie wapnia podczas stosowania produktu.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Specjalne środki ostrozności
Teryparatyd może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u osób z czynną kamicą moczową. Livogiva należy stosować ostrożnie u osób z czynną lub niedawno przebytą kamicą moczową, ze względu na ryzyko zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu produktu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienia ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Specjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania produktu u pacjentów z umiarkowaną niewydolnością nerek (patrz punkt 4.2). Stosowanie u młodych dorosłych Dane dotyczące stosowania produktu leczniczego u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym są ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania produktu Livogiva. W przypadku zajścia w ciążę należy przerwać stosowanie produktu Livogiva. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Specjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Substancje pomocnicze Produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Interakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie teryparatydu i raloksyfenu lub hormonalnej terapii zastępczej nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Wpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania produktu Livogiva, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania produktu Livogiva. Ciąża Stosowanie produktu Livogiva jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu Livogiva jest przeciwwskazane podczas karmienia piersią (patrz punkt 4.3). Nie wiadomo, czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ produktu na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Wpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Produkt leczniczy Livogiva nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów mechanicznych i obsługiwać urządzeń mechanicznych do czasu ustąpienia tych objawów.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Działania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi występującymi podczas stosowania teryparatydu są nudności, bóle kończyn, bóle i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8% pacjentów otrzymujących teryparatyd i u 84,5% pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W tabeli poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1 000), bardzo rzadko (<1/10 000). Tabela 1. Działania niepożądane
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Działania niepożądane
Klasyfikacja układów i narządówMedDRA Częstość występowania Działania niepożądane Zaburzenia krwi i układu chłonnego Często Niedokrwistość Zaburzenia układu immunologicznego Rzadko Anafilaksja Zaburzenia metabolizmu i odżywiania Często Hipercholesterolemia Niezbyt często Hiperkalcemia większa niż2,76 mmol/L, hiperurykemia Rzadko Hiperkalcemia większa niż3,25 mmol/L Zaburzenia psychiczne Często Depresja Zaburzenia układu nerwowego Często Zawroty głowy, ból głowy, rwa kulszowa, omdlenie Zaburzenia ucha i błędnika Często Zawroty głowy (spowodowane zaburzeniami błędnika) Zaburzenia serca Często Kołatanie serca Niezbyt często Tachykardia Zaburzenia naczyniowe Często Niedociśnienie Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia Często Duszność Niezbyt często Rozedma płuc Zaburzenia żołądka i jelit Często Nudności, wymioty, przepuklina rozworu przełykowego, chorobarefluksowa przełyku Niezbyt często Guzki krwawnicze Zaburzenia skóry i tkanki podskórnej Często Zwiększona potliwość Zaburzenia mięśniowo-szkieletowe i tkanki łącznej Bardzo często Ból kończyn Często Kurcze mięśni Niezbyt często Ból mięśni, ból stawów, kurczelub ból* mięśni pleców Zaburzenia nerek i dróg moczowych Niezbyt często Nietrzymanie moczu, nadmiernewydzielanie moczu, nagłe parcie na pęcherz, kamica nerkowa Rzadko Niewydolność nerek lub zaburzenia czynności nerek Zaburzenia ogólne i stany w miejscupodania Często Zmęczenie, ból w klatce piersiowej, osłabienie, łagodnei przemijające objawy w miejscupodania, w tym ból, obrzęk, rumień, miejscowe zasinienie, - CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Działania niepożądane
świąd i niewielkie krwawieniew miejscu wstrzyknięcia. Niezbyt często Rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia Rzadko Możliwe reakcje alergicznew krótkim czasie po wstrzyknięciu: ostre zaburzenia oddychania (duszność), obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowej, obrzęki (głównieobwodowe) Badania diagnostyczne Niezbyt często Zwiększenie masy ciała, szmery sercowe, zwiększenie aktywnościfosfatazy zasadowej - CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Działania niepożądane
* Silne kurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1% większą w porównaniu z placebo: zawroty głowy (spowodowane zaburzeniami błędnika), nudności, bóle kończyn, zawroty głowy, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8% pacjentów stosujących teryparatyd i 0,7% osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. Obserwowano przeciwciała przeciwko lekowi zgodne z przeciwciałami obserwowanymi w przypadku stosowania innych produktów leczniczych zawierających teryparatyd.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Działania niepożądane
Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Przedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu: ujawniająca się po pewnym czasie hiperkalcemia, ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania produktu na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu leczniczego, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 µg). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie lub ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Przedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na teryparatyd. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05AA02. Produkt leczniczy Livogiva jest biopodobnym produktem leczniczym. Szczegółowe informacje są dostępne na stronie internetowej Europejskiej Agencji ds. Leków http://www.ema.europa.eu. Mechanizm działania: Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rhPTH(1– 34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości poprzez bezpośredni wpływ na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększenie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki. Rezultat działania farmakodynamicznego Teryparatyd wspomaga proces tworzenia się kości.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Jest stosowany w leczeniu osteoporozy. Wpływ teryparatydu na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy. Podawanie teryparatydu raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej, dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna i bezpieczeństwo stosowania Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski wskaźnik masy ciała (BMI). Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości [np.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
mała gęstość mineralna kości (np. wskaźnik T score ≤−2), długotrwałe leczenie glikokortykosteroidami w dużych dawkach (np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy), choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych]. Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90% pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T score= -2,6 SD). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 IU witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 1). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy. Tabela 2.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Częstość występowania złamań u kobiet w okresie pomenopauzalnym
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Placebo(N = 544) (%) Teryparatyd(N = 541) (%) Ryzyko względne (95% CI)w porównaniu z placebo Nowe złamania kręgów 14,3 5,0b 0,35 a(≥1) (0,22; 0,55) Wielokrotne złamania 4,9 1,1b 0,23 akręgów (≥2) (0,09; 0,60) Złamania pozakręgowe spowodowane zwiększonąłamliwościąc 5,5% d2,6% 0,47(0,25; 0,87) Poważne złamania pozakręgowespowodowane zwiększoną 3,9% d1,5% 0,38(0,17; 0,86) łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber i miednicy) - CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących teryparatyd, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p≤0,001 w porównaniu z placebo c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. d p≤0,025 w porównaniu z placebo Po średnio 19 miesiącach leczenia, odnotowano zwiększenie gęstości mineralnej tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9% i 4% w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu terapii teryparatydem, 1262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41% (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83% stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5%, 2,6% i 3,9%. W okresie pomiędzy 18. a 24.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4%, 1,2% i 1,6%. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy 4 z losowym doborem uczestników, wzięło udział 1360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9% pacjentek, a 18,8% podczas badania przyjmowało jednocześnie glikokortykoidy. Ukończyło 24-miesięczną obserwację 1013 (74,5%) pacjentek.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykoidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1433 IU/dobę (1400 IU/dobę), a w grupie przyjmującej ryzedronian 1191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano radiografię kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4%) u pacjentek leczonych teryparatydem i 64/533 (12,0%) u pacjentek leczonych ryzedronianem, ryzyko względne (95% CI) = 0,44 (0,29-0,68), P <0,0001. Skumulowana częstość występowania łącznych złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8% w grupie pacjentek leczonych teryparatydem i 9,8% w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95% CI) = 0,48 (0,32–0,74), P=0,0009.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą w wyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia gęstość mineralna tkanki kostnej kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-score wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35% pacjentów miało złamania kręgów a 59% złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 IU witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5% i 1% w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18 miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg na dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28% pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 IU witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9% pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24% pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69% pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2%) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4%) (p<0,001).
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6%) w porównaniu z alendronianem (2,2%) (p<0,01), jak również szyjki kości udowej (3,7%) w porównaniu z alendronianem (2,1%) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7%, 0,9% i 0,4%. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7%) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7%) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0%) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5%) leczonej teryparatydem (p=0,84).
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2% w porównaniu −1,9%; p<0,001, dla kości biodra (3,8% w porównaniu 0,9%; p=0,005). Nie wykazano istotnego wpływu na częstość złamań kości.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Właściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 L/kg mc. Okres półtrwania teryparatydu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 L/h u kobiet i 94 L/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Przedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Produkt leczniczy nie wykazywał działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi, w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie poprzez mechanizm epigenetyczny.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Przedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. Nie stwierdzono guzów kości u operacyjnie pozbawionych jajników małp w okresie podawania przez 18 miesięcy ani w 3-letnim okresie obserwacji po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Dane farmaceutyczne
6. DANE FARMACEUTYCZNE: 6.1 Wykaz substancji pomocniczych Kwas octowy lodowaty Sodu octan trójwodny Mannitol, Metakrezol Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 2 lata Wykazano trwałość chemiczną, fizyczną i mikrobiologiczną stosowanego produktu w okresie 28 dni w temperaturze 2-8ºC. Po otwarciu produkt leczniczy można przechowywać nie dłużej niż 28 dni w temperaturze 2ºC do 8ºC. Za inne warunki i czas przechowywania stosowanego produktu odpowiedzialność ponosi użytkownik. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2ºC-8ºC). Bezpośrednio po każdym użyciu wstrzykiwacz należy ponownie umieścić w lodówce. Nie zamrażać. Nie przechowywać wstrzykiwacza z zamocowaną igłą. Po użyciu wstrzykiwacz zawsze przechowywać z nałożoną białą zatyczką, w celu ochrony przed światłem.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Dane farmaceutyczne
6.5 Rodzaj i zawartość opakowania Roztwór 2,7 mL we wkładzie (szkło silikonowane typu I) z jednej strony zamkniętym tłokiem z gumy bromobutylowej, a z drugiej strony złożonym dwuwarstwowym kapslem (laminat poliizopren/ guma bromobutylowa z aluminiową warstwą zewnętrzną). Wkłady są integralną i niewymienialną częścią wstrzykiwacza. Wstrzykiwacz składa się z przezroczystej obudowy mieszczącej wkład, białej zatyczki ochronnej do zakrycia obudowy mieszczącej wkład oraz trzonu wstrzykiwacza z czarnym przycisk podania dawki. Produkt Livogiva jest dostępny w opakowaniach zawierających 1 lub 3 wstrzykiwacze półautomatyczne napełnione. Jeden wstrzykiwacz półautomatyczny napełniony zawiera 28 dawek po 20 mikrogramów każda (w 80 mikrolitrach). Nie wszystkie wielkości opakowań muszą się znajdować w obrocie. 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Wstrzykiwacz przeznaczony jest do stosowania tylko przez jednego pacjenta.
- CHPL leku Livogiva, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mcl (1 dawka)Dane farmaceutyczne
Do każdego wstrzyknięcia musi być użyta nowa jałowa igła. Do produktu leczniczego nie dołączono igieł. Wstrzykiwacz można stosować z igłami przeznaczonymi do wstrzykiwaczy z insuliną. Po każdym wstrzyknięciu, wstrzykiwacz należy ponownie umieścić w lodówce. Nie należy stosować produktu Livogiva, jeżeli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Teriparatide SUN, 20 mikrogramów/80 mikrolitrów, roztwór do wstrzykiwań we wstrzykiwaczu 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu. Jeden wstrzykiwacz 2,4 ml zawiera 600 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań. Bezbarwny, przezroczysty roztwór. pH wynosi od 3,8 do 4,5. Osmolalność wynosi od 250 do 350 mOsmol.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWskazania do stosowania
4.1 Wskazania do stosowania Teriparatide SUN jest wskazany dla dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet po menopauzie wykazano istotne zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy spowodowanej długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn, o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecaną dawką produktu leczniczego Teriparatide SUN jest 20 mikrogramów, podawane raz na dobę. Całkowity maksymalny czas leczenia produktem Teriparatide SUN wynosi 24 miesiące (patrz punkt 4.4). Przez całe życie u pacjenta nie należy powtarzać 24 miesięcznego okresu leczenia produktem Teriparatide SUN. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Po zakończeniu terapii teryparatydem, pacjenci mogą stosować inne metody leczenia osteoporozy. Szczególne grupy pacjentów Pacjenci w podeszłym wieku Dostosowanie dawki w zależności od wieku nie jest wymagane (patrz punkt 5.2). Pacjenci z zaburzeniami czynności nerek Nie stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując produkt leczniczy u pacjentów z umiarkowanymi zaburzeniami czynności nerek.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDawkowanie
Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek. Pacjenci z zaburzeniami czynności wątroby Nie ma danych dotyczących stosowania produktu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność stosując teryparatyd. Dzieci, młodzież i młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Nie należy go stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Sposób podania Teriparatide SUN należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci muszą być poinformowani o właściwym sposobie wykonywania wstrzyknięcia (patrz punkt 6.6). Informacje dla pacjentów dotyczące prawidłowego sposobu użycia wstrzykiwacza dostępne są także w Instrukcji użycia.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1 ciąża i karmienie piersią (patrz punkty 4.4 i 4.6) wcześniej ujawniona hiperkalcemia ciężka niewydolność nerek metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie stan po radioterapii zewnętrznej lub wewnętrznej kośćca pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania produktu i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki teryparatydu . Nie jest konieczne rutynowe monitorowanie wapnia podczas stosowania produktu.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
Teryparatyd może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u osób z czynną kamicą moczową. Teryparatyd należy stosować ostrożnie u osób z czynną lub niedawno przebytą kamicą moczową, ze względu na ryzyko zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających badaniach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w ciągu 4 godzin po podaniu produktu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienia ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania produktu u pacjentów z umiarkowaną niewydolnością nerek. Stosowanie u młodych dorosłych Dane dotyczące stosowania produktu leczniczego u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym są ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania teryparatydu. W przypadku zajścia w ciążę należy przerwać stosowanie teryparatydu. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclSpecjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Zawartość sodu Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, to znaczy produkt uznaje się za „wolny od sodu”.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie teryparatydu i raloksyfenu lub hormonalnej terapii zastępczej nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym / Metody zapobiegania ciąży u kobiet W czasie stosowania teryparatydu, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania teryparatydu. Ciąża Stosowanie produktu Teriparatide SUN jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu Teriparatide SUN jest przeciwwskazane podczas karmienia piersią. Nie wiadomo czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ produktu na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Teriparatide SUN nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów mechanicznych i obsługiwać urządzeń mechanicznych do czasu ustąpienia tych objawów.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami występującymi u pacjentów leczonych teryparatydem są nudności, bóle kończyn, ból i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8 % pacjentów otrzymujących produkt leczniczy Teriparatide SUN i u 84,5 % pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W tabeli poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do 1/10), niezbyt często (≥1/1 000 do 1/100), rzadko (≥1/10 000 do 1/1 000), bardzo rzadko ( 1/10 000). Tabela 1 Częstość występowania złamań u kobiet po menopauzie
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Klasyfikacja MedDRA układów i organów Działanie niepożądane Częstotliwość Zaburzenia krwi i układu chłonnego anemia często Zaburzenia układu immunologicznego anafilaksja rzadko Zaburzenia metabolizmu i odżywiania hipercholesterolemia często hiperkalcemia większa niż 2,76 mmol/l, hiperurykemia niezbyt często hiperkalcemia większa niż 3,25mmol/l rzadko Zaburzenia psychiczne depresja często Zaburzenia układu nerwowego zawroty głowy, ból głowy, rwa kulszowa, omdlenie często Zaburzenia ucha i błędnika zawroty głowy pochodzeniabłędnikowego często Zaburzenia serca kołatanie serca często tachykardia niezbyt często Zaburzenia naczyniowe niedociśnienie często Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia duszność często rozedma płuc niezbyt często Zaburzenia żołądka i jelit nudności, wymioty, przepuklina rozworu przełykowego, choroba refluksowa przełyku często guzy krwawnicze niezbyt często Zaburzenia skóry i tkankipodskórnej zwiększona potliwość często Zaburzenia mięśniowo- szkieletowe i tkanki łącznej ból kończyn bardzo często kurcze mięśni często ból mięśni, ból stawów, kurcze lub ból* mięśni pleców niezbyt często Zaburzenia nerek i dróg moczowych nietrzymanie moczu, nadmierne wydzielanie moczu, nagłe parcie na pęcherz, kamica nerkowa niezbyt często niewydolność nerek lubzaburzenia czynności nerek rzadko - CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Klasyfikacja MedDRA układów i organów Działanie niepożądane Częstotliwość Zaburzenia ogólne i stany w miejscu podania zmęczenie, ból w klatce piersiowej, osłabienie, łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień, miejscowe zasinienie, świąd i niewielkie krwawienie w miejscuwstrzyknięcia często rumień w miejscu wstrzyknięcia, reakcja wmiejscu wstrzyknięcia niezbyt często możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostre zaburzenia oddychania (duszność), obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból wklatce piersiowej, obrzęki (głównie obwodowe) rzadko Badania diagnostyczne zwiększenie masy ciała, szmery sercowe, zwiększenie aktywności fosfatazyzasadowej niezbyt często - CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
* Silne kurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1 % większą w porównaniu z placebo: zawroty głowy (spowodowane zaburzeniami błędnika), nudności, bóle kończyn, zawroty głowy, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8 % pacjentów stosujących teryparatyd i 0,7 % osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. W dużym badaniu klinicznym, u 2,8 % kobiet otrzymujących teryparatyd wykryto przeciwciała reagujące krzyżowo z teryparatydem. Przeciwciała zazwyczaj wykrywano po 12 miesiącach leczenia, a ich miano zmniejszało się po odstawieniu produktu.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDziałania niepożądane
Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu: ujawniająca się po pewnym czasie hiperkalcemia, ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania produktu na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu: Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 800 g). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie lub ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na produkt Teriparatide SUN. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05 AA02 Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rhPTH(1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki. Rezultat działania farmakodynamicznego Teryparatyd wspomaga proces tworzenia się kości. Jest stosowany w leczeniu osteoporozy. Wpływ produktu Teriparatide SUN na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podawanie Teriparatide SUN raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI). Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np. mała gęstość mineralna kości [np.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90 % pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T-score= -2,6 SD). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 IU witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do 24 miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 1). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Tabela 2 Częstość występowania złamań u kobiet w okresie pomenopauzalnym
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Placebo (N = 544) (%) Teryparatyd(N = 541) (%) Ryzyko względne (95% CI)w porównaniu zplacebo Nowe złamania kręgów 14,3 5,0 b 0,35 (1)a (0,22; 0,55) Wielokrotne złamania 4,9 1,1 b 0,23 kręgów (2)a (0,09; 0,60) Złamania pozakręgowe spowodowane zwiększonąłamliwością c 5,5 2,6d 0,47(0,25; 0,87) Poważne złamania pozakręgowe spowodowane zwiększoną łamliwością (szyjki kości udowej, kości promieniowej, kości ramienia, żeber imiednicy) 3,9 1,5d 0,38(0,17; 0,86) - CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących Teriparatide SUN, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p 0,001 w porównaniu z placebo c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. d p 0,025 w porównaniu z placebo Po średnio 19 miesiącach leczenia, odnotowano zwiększenie gęstości mineralnej tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9 % i 4 % w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu terapii teryparatydem, 1262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. Średnio w okresie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41 % (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w ciągu ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83 % stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5 %, 2,6 % i 3,9 %. W okresie pomiędzy 18. a 24.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4 %, 1,2 % i 1,6 %. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy 4 z losowym doborem uczestników, wzięło udział 1360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9% pacjentek, a 18,8% podczas badania przyjmowało jednocześnie glikokortykoidy. Ukończyło 24-miesięczną obserwację 1013 (74,5%) pacjentek.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykoidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1433 IU/dobę (1400 IU/dobę), a w grupie przyjmującej ryzedronian 1191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano radiografię kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4%) u pacjentek leczonych teryparatydem i 64/533 (12,0%) u pacjentek leczonych ryzedronianem, ryzyko względne (95% CI) = 0,44 (0,29-0,68), P <0,0001. Skumulowana częstość występowania łącznych złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8% w grupie pacjentek leczonych teryparatydem i 9,8% w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95% CI) = 0,48 (0,32-0,74), P=0,0009.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą wwyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia gęstość mineralna tkanki kostnej kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35 % pacjentów miało złamania kręgów a 59 % złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 IU witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w ciągu trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5 % i 1 % w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18 miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg na dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 IU witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69 % pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2 %) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4 %) (p<0,001).
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6 %) w porównaniu z alendronianem (2,2 %) (p<0,01), jak również szyjki kości udowej (3,7 %) w porównaniu z alendronianem (2,1 %) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7 %, 0,9 % i 0,4 %. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7 %) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7 %) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0 %) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5 %) leczonej teryparatydem (p=0,84).
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2 % w porównaniu −1,9 %; p<0,001, dla kości biodra (3,8 % w porównaniu 0,9 %; p=0,005). Nie wykazano istotnego wpływu na częstość złamań kości.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania produktu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31-85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Produkt leczniczy nie wykazywał działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
6. DANE FARMACEUTYCZNE: 6.1 Wykaz substancji pomocniczych Kwas octowy lodowaty (E 260) Octan sodu bezwodny (E 262) Mannitol (E 421) Metakrezol Kwas solny (do ustalenia pH) (E507) Wodorotlenek sodu (do ustalenia pH) (E524) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 2 lata Okres ważności po pierwszym otwarciu: Wykazano trwałość chemiczną, fizyczną i mikrobiologiczną stosowanego produktu w okresie 28 dni w temperaturze 2-8ºC. Po otwarciu produkt leczniczy można przechowywać nie dłużej niż 28 dni w temperaturze 2ºC do 8ºC. Za inne warunki i czas przechowywania stosowanego produktu odpowiedzialność ponosi użytkownik. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2ºC – 8ºC). Bezpośrednio po każdym użyciu wstrzykiwacz należy ponownie umieścić w lodówce. Nie zamrażać.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
Warunki przechowywania po pierwszym otwarciu produktu leczniczego, patrz punkt 6.3. 6.5 Rodzaj i zawartość opakowania Roztwór 2,4 ml we wkładzie (szkło silikonowane) zamkniętym korkiem (z gumy halobutylowej), zatyczką (poliizopren/ laminat z gumy bromobutylowej/ aluminium) umieszczony w jednorazowym wstrzykiwaczu. Teriparatide SUN jest dostępny w opakowaniach zawierających 1 lub 3 wstrzykiwacze. Jeden wstrzykiwacz zawiera 28 dawek po 20 mikrogramów każda (w 80 mikrolitrach). Nie wszystkie wielkości opakowań muszą się znajdować w obrocie. 6.6 Specjalne środki ostrożności dotyczące usuwania Postępowanie ze wtrzykiwaczem Teriparatide SUN znajduje się w jednorazowym wstrzykiwaczu. Przeznaczony jest do stosowania przez jednego pacjenta. Do każdego wstrzyknięcia musi być użyta nowa jałowa igła (31 Gauge, o długości 5 mm). Do wstrzykiwaczy nie dołączono igieł. Po każdym wstrzyknięciu, wstrzykiwacz należy ponownie umieścić w lodówce. Nie należy przechowywać ampułkostrzykawki z dołączoną igłą.
- CHPL leku Teriparatide SUN, roztwór do wstrzykiwań we wstrzykiwaczu, 20 mcg/80 mclDane farmaceutyczne
Nie należy stosować produktu Teriparatide SUN, jeżeli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Usuwanie Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Kauliv 20 mikrogramów/80 mikrolitrów, roztwór do wstrzykiwań 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Jedna dawka 80 mikrolitrów zawiera 20 mikrogramów teryparatydu*. Każdy wkład z 3 ml roztworu zawiera 750 mikrogramów teryparatydu (co odpowiada 250 mikrogramom na mililitr). *Teryparatyd, rhPTH(1-34), wytwarzany metodą rekombinacji DNA przez E.coli , ma strukturę identyczną z sekwencją 34 N-końcowych aminokwasów endogennego ludzkiego parathormonu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań. Bezbarwny, przezroczysty roztwór do wstrzykiwań.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Kauliv jest wskazany dla pacjentów dorosłych. Leczenie osteoporozy u kobiet w okresie pomenopauzalnym i u mężczyzn o podwyższonym ryzyku złamań (patrz punkt 5.1). U kobiet w okresie pomenopauzalnym wykazano znaczące zmniejszenie częstości występowania złamań kręgów oraz złamań pozakręgowych, nie dotyczy to jednak szyjki kości udowej. Leczenie osteoporozy związanej z długotrwałym stosowaniem glikokortykosteroidów o działaniu ogólnoustrojowym u kobiet i mężczyzn, o podwyższonym ryzyku złamań (patrz punkt 5.1).
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecana dawka produktu Kauliv to 20 mikrogramów, podawane jeden raz na dobę. Jeżeli zawartość wapnia i witaminy D w diecie nie jest wystarczająca, należy ją uzupełniać stosując preparaty zawierające wapń i witaminę D. Całkowity maksymalny czas leczenia teryparatydem wynosi 24 miesiące (patrz punkt 4.4). Przez całe życie u pacjenta nie należy powtarzać 24 miesięcznego okresu leczenia teryparatydem. Po zakończeniu leczenia teryparatydem, pacjenci mogą stosować inne metody leczenia osteoporozy. Populacje szczególne Pacjenci w podeszłym wieku Nie jest konieczna modyfikacja dawki produktu zależnie od wieku pacjenta (patrz punkt 5.2). Zaburzenia czynności nerek Nie wolno stosować teryparatydu u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.3). Należy zachować ostrożność stosując teryparatyd u pacjentów z umiarkowanymi zaburzeniami czynności nerek.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
Nie jest wymagane zachowanie szczególnej ostrożności u pacjentów z łagodnymi zaburzeniami czynności nerek. Zaburzenia czynności wątroby Nie ma danych dotyczących stosowania teryparatydu u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.3). Z tego względu należy zachować ostrożność podczas stosowania teryparatydu. Dzieci i młodzież oraz młodzi dorośli, przed zakończeniem rozwoju nasad kości długich Nie ustalono bezpieczeństwa i skuteczności teryparatydu u dzieci i młodzieży w wieku poniżej 18 lat. Teryparatydu nie należy stosować u dzieci i młodzieży (wiek poniżej 18 lat) oraz u młodych dorosłych, przed zakończeniem rozwoju nasad kości długich. Sposób podawania Produkt leczniczy Kauliv należy podawać raz na dobę we wstrzyknięciu podskórnym w udo lub brzuch. Pacjenci powinni zostać przeszkoleni w zakresie właściwego sposobu wykonywania wstrzyknięcia (patrz punkt 6.6). W celu zapoznania się z instrukcjami dotyczącymi produktu leczniczego przed podaniem (patrz punkt 6.6).
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDawkowanie
Instrukcje użytkowania, dołączone do opakowania wstrzykiwacza, także mogą być stosowane do poinstruowania pacjentów jak prawidłowo stosować wstrzykiwacz.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclPrzeciwwskazania
4.3 Przeciwwskazania Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1. Ciąża i karmienie piersią (patrz punkty 4.4 i 4.6). Wcześniej ujawniona hiperkalcemia. Ciężkie zaburzenia czynności nerek. Metaboliczne choroby kości (w tym nadczynność przytarczyc i choroba Pageta kości), z wyjątkiem pierwotnej osteoporozy i osteoporozy spowodowanej stosowaniem glikokortykosteroidów. Zwiększenie aktywności fosfatazy zasadowej, o niewyjaśnionej przyczynie. Stan po radioterapii zewnętrznej lub wewnętrznej kośćca. Pacjenci z nowotworami złośliwymi układu kostno-szkieletowego lub przerzutami do kości nie powinni być leczeni teryparatydem.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Identyfikowalność W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu. Stężenie wapnia w surowicy i w moczu U osób z prawidłowym stężeniem wapnia we krwi po wstrzyknięciu teryparatydu obserwowano niewielkie i przemijające zwiększenie stężenia wapnia w surowicy krwi. Maksymalne stężenie wapnia w surowicy krwi występowało po 4-6 godzinach od podania i powracało do wartości wyjściowych po 16-24 godzinach od podania teryparatydu. Z tego powodu próbkę krwi do badania stężenia wapnia w surowicy krwi, należy pobrać od pacjenta co najmniej 16 godzin po wstrzyknięciu ostatniej dawki teryparatydu. Nie jest konieczne rutynowe monitorowanie wapnia podczas leczenia.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
Teryparatyd może powodować niewielkie zwiększenie wydalania wapnia z moczem, jednak w badaniach klinicznych częstość występowania nadmiernego wydalania wapnia z moczem u pacjentów przyjmujących teryparatyd nie różniła się od obserwowanej u pacjentów otrzymujących placebo. Kamica moczowa Nie przeprowadzano badań dotyczących stosowania teryparatydu u pacjentów z czynną kamicą moczową. Teryparatyd należy stosować ostrożnie u pacjentów z czynną lub niedawno przebytą kamicą moczową, ze względu na możliwość zaostrzenia przebiegu tej choroby. Niedociśnienie ortostatyczne W krótko trwających próbach klinicznych z zastosowaniem teryparatydu obserwowano pojedyncze przypadki przemijającego niedociśnienia ortostatycznego. Zazwyczaj niedociśnienie ortostatyczne występowało w przez 4 godzin po podaniu i ustępowało samoistnie po kilku minutach lub godzinach. Przemijające niedociśnienie ortostatyczne występowało podczas podawania kilku pierwszych dawek produktu.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
Nie uniemożliwiało to kontynuowania leczenia. Ułożenie pacjenta w pozycji półleżącej łagodziło objawy. Zaburzenia czynności nerek Należy zachować ostrożność podczas stosowania u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Stosowanie u młodych dorosłych Doświadczenie związane ze stosowaniem u młodych dorosłych, w tym u kobiet w okresie przedmenopauzalnym, jest ograniczone (patrz punkt 5.1). W tej populacji leczenie należy zastosować tylko jeśli spodziewane korzyści wyraźnie przewyższają ryzyko. Kobiety w wieku rozrodczym muszą stosować skuteczną metodę zapobiegania ciąży w trakcie stosowania teryparatydu. W przypadku zajścia w ciążę należy przerwać stosowanie teryparatydu. Czas trwania leczenia Wyniki badań przeprowadzonych na szczurach wskazują na zwiększoną częstość występowania kostniakomięsaka podczas długotrwałego stosowania teryparatydu (patrz punkt 5.3). Nie należy przekraczać zalecanego maksymalnego okresu leczenia, tj.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclSpecjalne środki ostrozności
24 miesięcy, do czasu uzyskania nowych danych klinicznych. Substancja pomocnicza Lek zawiera mniej niż 1 mmol (2 mg) sodu na jednostkę dawkowania, to znaczy lek uznaje się za „wolny od sodu”.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji W badaniu obejmującym 15 zdrowych osób, którym codziennie podawano digoksynę, aż do osiągnięcia stanu równowagi stężeń, zastosowanie pojedynczej dawki teryparatydu nie zmieniało wpływu digoksyny na serce. Z opisów sporadycznych przypadków wynika jednak, że hiperkalcemia może być czynnikiem predysponującym do wystąpienia działania toksycznego glikozydów naparstnicy. Ze względu na to, że teryparatyd powoduje przemijające zwiększenie stężenia wapnia w surowicy krwi, należy stosować go ostrożnie u osób przyjmujących glikozydy naparstnicy. Badano farmakodynamiczne interakcje teryparatydu i hydrochlorotiazydu. Nie odnotowano żadnych klinicznie istotnych interakcji. Jednoczesne stosowanie raloksyfenu lub hormonalnej terapii zastępczej z teryparatydem nie zmieniało wpływu teryparatydu na stężenie wapnia w surowicy krwi lub w moczu ani na występowanie istotnych klinicznie działań niepożądanych.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Kobiety w wieku rozrodczym/Metody zapobiegania ciąży u kobiet W czasie stosowania teryparatydu, kobiety w wieku rozrodczym powinny stosować skuteczną metodę zapobiegania ciąży. W przypadku zajścia w ciążę, należy zaprzestać stosowania teryparatydu. Ciąża Stosowanie produktu Kauliv jest przeciwwskazane w okresie ciąży (patrz punkt 4.3). Karmienie piersią Stosowanie produktu Kauliv jest przeciwwskazane podczas karmienia piersią. Nie wiadomo, czy teryparatyd przenika do mleka kobiecego. Płodność W badaniach na królikach wykazano toksyczny wpływ na reprodukcję (patrz punkt 5.3). Nie badano wpływu teryparatydu na rozwój ludzkiego płodu. Potencjalne zagrożenie dla człowieka nie jest znane.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Produkt Kauliv nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. U niektórych pacjentów obserwowano przemijające niedociśnienie ortostatyczne oraz zawroty głowy. Takie osoby nie powinny prowadzić pojazdów i obsługiwać maszyn do czasu ustąpienia tych objawów.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi występującymi podczas stosowania teryparatydu są nudności, bóle kończyn, bóle i zawroty głowy. Tabelaryczne zestawienie działań niepożądanych Podczas badań z zastosowaniem teryparatydu u 82,8 % pacjentów otrzymujących teryparatyd i u 84,5 % pacjentów przyjmujących placebo wystąpiło co najmniej jedno zdarzenie niepożądane. W tabeli poniżej podano działania niepożądane zgłaszane po zastosowaniu teryparatydu w badaniach klinicznych dotyczących leczenia osteoporozy oraz po wprowadzeniu produktu do obrotu. W celu oszacowania częstości występowania działań niepożądanych zastosowano następującą klasyfikację: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1000 do <1/100) oraz rzadko (≥1/10 000 do <1/1000). Tabela 1. Działania niepożądane
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Klasyfikacja układów Częstość Działania niepożądane - CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
i narządów MedDRA Zaburzenia krwi iukładu chłonnego często niedokrwistość Zaburzenia układu immunologicznego Rzadko Anafilaksja Zaburzenia metabolizmu i odżywania często hipercholesterolemia niezbyt często hiperkalcemia powyżej 2,76 mmol/l, hiperurykemia rzadko hiperkalcemia powyżej 3,25 mmol/l Zaburzeniapsychiczne często depresja Zaburzenia układu nerwowego często zawroty głowy, ból głowy, rwa kulszowa, omdlenia Zaburzenia ucha i błędnika często zawroty głowy Zaburzenia serca często kołatanie serca niezbyt często tachykardia Zaburzenia naczyniowe często niedociśnienie Zaburzenia układu oddechowego, klatki piersioweji śródpiersia często duszność niezbyt często rozedma płuc Zaburzenia żołądka ijelit często nudności, wymioty, przepuklina rozworu przełykowaegorefluks żołądkowo-przełykowy niezbyt często hemoroidy Zaburzenia skóry i tkanki podskórnej często wzmożone pocenie się Zaburzeniamięśniowo-szkieletowei tkanki łącznej bardzo często bóle kończyn często skurcze mięśni niezbyt często bóle mięśni, ból stawów, skurcze/ból* mięśni pleców Zaburzenia nerek iukładu moczowego niezbyt często nietrzymanie moczunagłe parcie na pęcherz, kamica nerkowa rzadko niewydolność/zaburzenia czynności nerek Zaburzenia ogólne i stany w miejscu podania często zmęczenie, ból w klatce piersiowej, osłabienie łagodne i przemijające objawy w miejscu podania, w tym ból, obrzęk, rumień miejscowe zasinienie, świąd iniewielkie krwawienie w miejscu wstrzyknięcia - CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
niezbyt często rumień w miejscu wstrzyknięcia, reakcja w miejscu wstrzyknięcia rzadko możliwe reakcje alergiczne w krótkim czasie po wstrzyknięciu: ostra duszność, obrzęk w okolicy ust i twarzy, pokrzywka uogólniona, ból w klatce piersiowejobrzęki (głównie obwodowe) Badaniadiagnostyczne niezbyt często zwiększenie masy ciała. szmery sercowezwiększenie aktywności fosfatazy alkalicznej - CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
* Silne skurcze lub ból mięśni pleców zgłaszano po upływie kilku minut po wstrzyknięciu. Opis wybranych działań niepożądanych W badaniach klinicznych następujące działania niepożądane były zgłaszane z częstością ≥ 1 % większą w porównaniu z placebo: zawroty głowy pochodzenia błędnikowego, nudności, bóle kończyn, zawroty głowy pochodzenia ośrodkowego, depresja, duszność. Teryparatyd powoduje zwiększenie stężenia kwasu moczowego w surowicy krwi. Podczas badań klinicznych u 2,8 % pacjentów stosujących teryparatyd i 0,7 % osób przyjmujących placebo stężenie kwasu moczowego przekraczało górną granicę zakresu wartości przyjętych za prawidłowe. Hiperurykemia nie powodowała jednak zwiększenia częstości występowania dny, bólów stawów ani kamicy układu moczowego. Przeciwciała przeciwlekowe , jeśli występują, są na ogół obserwowane przy stosowaniu innych produktów leczniczych zawierających teryparatyd.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDziałania niepożądane
Nie stwierdzono reakcji nadwrażliwości, reakcji alergicznych, zmian stężenia wapnia w surowicy krwi lub wpływu produktu na gęstość mineralną tkanki kostnej (BMD). Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe Teryparatyd podawano w dawkach pojedynczych do 100 mikrogramów, oraz w dawkach wielokrotnych do 60 mikrogramów na dobę przez 6 tygodni. Objawy, których można się spodziewać po przedawkowaniu obejmują ujawniającą się po pewnym czasie hiperkalcemię oraz ryzyko wystąpienia niedociśnienia ortostatycznego. Mogą także wystąpić nudności, wymioty, zawroty i bóle głowy. Przypadki przedawkowania na podstawie spontanicznych doniesień zgłaszanych po wprowadzeniu produktu leczniczego do obrotu Po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki błędnego dawkowania produktu, polegające na jednorazowym podaniu całej zawartości wstrzykiwacza zawierającego teryparatyd (do 750 mikrogramów). Zgłaszano wystąpienie przemijających działań niepożądanych: nudności, osłabienie i (lub) ospałość i niedociśnienie tętnicze. W niektórych przypadkach przedawkowania produktu nie obserwowano żadnych działań niepożądanych.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedawkowanie
Nie zgłoszono ani jednego przypadku zgonu pacjenta w wyniku przedawkowania produktu leczniczego. Postępowanie w przypadku przedawkowania Nie istnieje swoista odtrutka na teryparatyd. Postępowanie w przypadku podejrzenia przedawkowania powinno obejmować krótkotrwałe odstawienie produktu, kontrolę stężenia wapnia w surowicy krwi oraz odpowiednie leczenie podtrzymujące, np. nawodnienie.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi, kod ATC: H05AA02 Kauliv jest produktem leczniczym biopodobnym. Szczegółowe informacje są dostępne na stronie internetowej Europejskiej Agencji Leków http://www.ema.europa.eu . Mechanizm działania Endogenny parathormon (PTH) zbudowany z 84 aminokwasów jest głównym czynnikiem regulującym metabolizm wapnia i fosforanów w tkance kostnej i w nerkach. Teryparatyd (rh PTH (1-34)) jest aktywnym fragmentem (1-34) endogennego ludzkiego parathormonu. Działanie fizjologiczne PTH obejmuje pobudzanie procesu tworzenia kości wpływając bezpośrednio na komórki kościotwórcze (osteoblasty), pośrednio powodując zwiększenie wchłaniania wapnia w jelitach oraz zwiększanie zwrotnego wchłaniania wapnia w kanalikach nerkowych i wydalania fosforanów przez nerki. Działania farmakodynamicznego Teryparatyd wspomaga proces tworzenia się kości i jest stosowany w leczeniu osteoporozy.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Wpływ teryparatydu na układ kostny zależy od przebiegu reakcji organizmu na produkt leczniczy. Podawanie teryparatydu raz na dobę zwiększa odkładanie się nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej dzięki większemu pobudzaniu aktywności osteoblastów niż osteoklastów. Skuteczność kliniczna i bezpieczeństwo stosowania Czynniki ryzyka W celu identyfikacji kobiet i mężczyzn o podwyższonym ryzyku osteoporotycznych złamań, którzy mogą odnieść korzyść z leczenia, należy rozważyć niezależne czynniki ryzyka takie jak mała gęstość mineralna kości (BMD), wiek, wcześniejsze złamania, złamania szyjki kości udowej u członków rodziny, zwiększona przebudowa kości i niski indeks masy ciała (BMI). Należy przyjąć, że wysokie ryzyko złamań kości dotyczy kobiet w okresie przedmenopauzalnym z osteoporozą spowodowaną stosowaniem glikokortykosteroidów, u których wystąpiło złamanie kości lub u których stwierdzono zespół czynników ryzyka predysponujących do zaliczenia do grupy wysokiego ryzyka złamań kości (np.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
mała gęstość mineralna kości [np. wskaźnik T score ≤−2], długotrwałe leczenie glikokortykosteroidami w dużych dawkach [np. ≥7,5 mg na dobę przez co najmniej 6 miesięcy], choroba podstawowa o dużej intensywności, mała aktywność hormonów płciowych). Osteoporoza w okresie pomenopauzalnym W głównym badaniu wzięło udział 1637 kobiet w okresie pomenopauzalnym (średnia wieku 69,5 lat). W punkcie wyjściowym badania 90 % pacjentek przebyło wcześniej jedno lub więcej złamań kręgów, a gęstość mineralna kości mierzona w kręgach wynosiła średnio BMD = 0,82 g/cm 2 (co odpowiadało wartości wskaźnika T-score= -2,6 SD). Wszystkim pacjentkom podawano 1000 mg wapnia na dobę i przynajmniej 400 j.m. witaminy D na dobę. Wyniki stosowania teryparatydu przez okres do miesięcy (średnio 19 miesięcy) wykazały statystycznie istotne zmniejszeniu częstości złamań (Tabela 2). Aby zapobiec nowym złamaniom (jednemu lub większej ilości nowych złamań) kręgów, 11 kobiet musiano leczyć średnio przez 19 miesięcy. Tabela 2.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Częstość występowania złamań u kobiet w wieku pomenopauzalnym
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Placebo(N = 544) (%) Teryparatyd(N = 541) (%) Ryzyko względna (95% CI) w porównaniu doplacebo Nowe złamania kręgów (≥1) a 14,3 5,0b 0,35(0,22, 0,55) Wielokrotne złamania kręgów (≥1) a 4,9 1,1b 0,23(0,09, 0,60) Złamania pozakręgowec spowodowane zwiększoną łamliwością c 5,5% 2,6%b 0,47(0,25, 0,87) Ciężkie złamania pozakręgowec spowodowane zwiększoną łamliwością (szyjki kości udowej, kościpromieniowej, kości ramienia, żeber imiednicy) 3,9% 1.5%b 0,38(0,17, 0,86) - CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Oznaczenia: N= liczba pacjentów losowo przypisanych do danej grupy leczenia; CI = przedział ufności. a Częstość występowania złamań kręgów była oceniana w grupie 448 pacjentów stosujących placebo i w grupie 444 pacjentów stosujących teryparatyd, u których wykonano zdjęcia rentgenowskie kręgosłupa w punkcie wyjściowym i w czasie badania. b p≤0,001 w porównaniu z placebo. c Nie stwierdzono istotnego zmniejszenia występowania złamań szyjki kości udowej. p≤0,025 w porównaniu z placebo. Po średnio 19 miesiącach leczenia, odnotowano zwiększenie BMD lędźwiowego odcinka kręgosłupa i kości biodra odpowiednio o 9 % i 4 % w porównaniu z placebo (p<0,0001). Postępowanie po leczeniu: Po zakończeniu leczenia teryparatydem, 1262 kobiet w okresie pomenopauzalnym, które uczestniczyły w badaniu kluczowym, włączono do badania obserwacyjnego. Podstawowym celem tego badania było zebranie danych dotyczących bezpieczeństwa stosowania teryparatydu.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Podczas badania obserwacyjnego pozwolono stosować inne metody leczenia osteoporozy i wykonywano dodatkową ocenę złamań kręgów. W okresie o medianie 18 miesięcy po zakończeniu stosowania teryparatydu odnotowano zmniejszenie o 41% (p=0,004) liczby pacjentek co najmniej z jednym nowym złamaniem kręgu w porównaniu z placebo. W otwartym badaniu 503 kobiety w okresie pomenopauzalnym z zaawansowaną osteoporozą, u których w przez ostatnich trzech latach wystąpiło złamanie spowodowane zwiększoną łamliwością kości (u 83 % stosowano wcześniej leczenie osteoporozy), były leczone teryparatydem w okresie do 24 miesięcy. Po 24 miesiącach średnie zwiększenie w odniesieniu do wartości wyjściowych gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 10,5%, 2,6% i 3,9%. W okresie pomiędzy 18. a 24.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
miesiącem leczenia średnie zwiększenie gęstości mineralnej tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjki kości udowej wynosiło odpowiednio 1,4 %, 1,2% i 1,6%. W trwającym 24 miesiące, podwójnie zaślepionym, kontrolowanym lekiem porównawczym badaniu fazy IV z losowym doborem uczestników, wzięło udział 1 360 kobiet w okresie pomenopauzalnym z rozpoznaną osteoporozą. Do grupy przyjmującej teryparatyd zostało losowo przydzielonych 680 pacjentek i 680 pacjentek zostało losowo przydzielonych do grupy przyjmującej doustnie ryzedronian w dawce 35 mg/tydzień. Wyjściowo średni wiek kobiet wynosił 72,1 lat, a mediana złamań kręgów wynosiła 2. Wcześniejsze leczenie bisfosfonianami otrzymało 57,9 % pacjentek, a 18,8 % podczas badania przyjmowało jednocześnie glikokortykosteroidy. 24-miesięczną obserwację ukończyło 1 013 (74,5 %) pacjentek.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Średnia (mediana) skumulowana dawka glukokortykosteroidu wynosiła 474,3 (66,2) mg w grupie stosującej teryparatyd i 898,0 (100,0) mg w grupie stosującej ryzedronian. Średnie (mediana) spożycie witaminy D w grupie przyjmującej teryparatyd wynosiło 1 433 IU/dobę (1 400 IU/dobę), a w grupie przyjmującej ryzedronian 1 191 IU/dobę (900 IU/dobę). W przypadku osób, u których wyjściowo i kontrolnie wykonano badanie rentgenowskie kręgosłupa, częstość występowania nowych złamań kręgów wynosiła 28/516 (5,4 %) u pacjentek leczonych teryparatydem i 64/533 (12,0 %) u pacjentek leczonych ryzedronianem, ryzyko względne (95 % CI) = 0,44 (0,29 - 0,68), p<0,0001. Łączna, skumulowana częstość występowania złamań klinicznych (kliniczne złamania kręgosłupa i inne) wynosiła 4,8 % w grupie pacjentek leczonych teryparatydem i 9,8 % w grupie pacjentek leczonych ryzedronianem, współczynnik ryzyka (95 % CI) = 0,48 (0,32- 0,74), p=0,0009.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza u mężczyzn W badaniu klinicznym brało udział 437 mężczyzn (średnia wieku 58,7 lat) z osteoporozą powstałą w wyniku niedoczynności gonad (stwierdzona w przypadku małego porannego stężenia wolnego testosteronu lub zwiększonego stężenia FSH lub LH) lub osteoporozą idiopatyczną. W punkcie wyjściowym średnia BMD kręgosłupa i szyjki kości udowej oznaczana za pomocą wskaźnika T-scores wynosiła odpowiednio -2,2 i -2,1. W punkcie wyjściowym 35 % pacjentów miało złamania kręgów a 59 % złamania pozakręgowe. Wszystkim uczestnikom podawano 1000 mg wapnia na dobę oraz co najmniej 400 j.m. witaminy D na dobę. Wskaźnik BMD (gęstości mineralnej tkanki kostnej) kręgosłupa lędźwiowego istotnie wzrósł w przez trzech miesięcy. Po 12 miesiącach leczenia odnotowano zwiększenie BMD odcinka lędźwiowego kręgosłupa i kości biodra odpowiednio o 5 % i 1 % w porównaniu z placebo. Nie stwierdzono jednak istotnego wpływu leczenia na częstość występowania złamań.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Osteoporoza spowodowana stosowaniem glikokortykosteroidów Skuteczność teryparatydu wykazano w pierwszej 18-miesięcznej fazie 36-miesięcznego randomizowanego kontrolowanego badania z podwójnie ślepą próbą, z użyciem produktu porównawczego (alendronian w dawce 10 mg/dobę) z udziałem mężczyzn i kobiet (N=428) długotrwale stosujących glikokortykosteroidy (w dawce odpowiadającej co najmniej 5 mg prednizonu przez przynajmniej 3 miesiące). W punkcie wyjściowym badania u 28 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Wszystkim pacjentom podawano 1000 mg wapnia na dobę i 800 j.m. witaminy D na dobę. W badaniu uczestniczyły kobiety w okresie pomenopauzalnym (N=277), kobiety w okresie przed menopauzą (N=67) i mężczyźni (N=83). W punkcie wyjściowym średni wiek kobiet w okresie pomenopauzalnym wynosił 61 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,7, mediana przyjmowanej dawki odpowiadała 7,5 mg prednizonu na dobę, i u 34 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
W punkcie wyjściowym średni wiek kobiet w okresie przed menopauzą wynosił 37 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,5, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, u 9 % pacjentek stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich; średni wiek mężczyzn wynosił 57 lat, średnia gęstość mineralna tkanki kostnej (BMD) lędźwiowego odcinka kręgosłupa oznaczana za pomocą wskaźnika T score wynosiła −2,2, mediana przyjmowanej dawki odpowiadała 10 mg prednizonu na dobę, i u 24 % pacjentów stwierdzono co najmniej jedno złamanie kręgu widoczne na zdjęciach rentgenowskich. Pierwszą fazę badania trwającą 18 miesięcy ukończyło 69 % pacjentów. W punkcie końcowym po 18 miesiącach wykazano, że stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej tkanki kostnej (7,2 %) odcinka lędźwiowego kręgosłupa w porównaniu z alendronianem (3,4 %) (p<0,001).
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
Stosowanie teryparatydu spowodowało istotne zwiększenie gęstości mineralnej kości biodra (3,6 %) w porównaniu z alendronianem (2,2 %) (p<0,01), jak również szyjki kości udowej (3,7 %) w porównaniu z alendronianem (2,1 %) (p<0,05). W okresie pomiędzy 18. a 24. miesiącem leczenia teryparatydem gęstość mineralna tkanki kostnej w odcinku lędźwiowym kręgosłupa, kości biodra i szyjce kości udowej dodatkowo zwiększyła się o odpowiednio o 1,7 %, 0,9 % i 0,4 %. Po 36 miesiącach analiza zdjęć rentgenowskich kręgosłupa 169 pacjentów leczonych alendronianem i 173 pacjentów stosujących teryparatyd wykazała, że u 13 pacjentów z grupy leczonej alendronianem (7,7 %) wystąpiło nowe złamanie kręgu, w porównaniu z 3 pacjentami z grupy leczonej teryparatydem (1,7 %) (p=0,01). Ponadto u 15 z 214 pacjentów leczonych alendronianem (7,0 %) wystąpiły złamania pozakręgowe w porównaniu z 16 pacjentami z grupy 214 osobowej (7,5 %) leczonej teryparatydem (p=0,84).
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakodynamiczne
U kobiet w okresie przed menopauzą zwiększenie gęstości mineralnej kości od punktu wyjściowego do końcowego po 18 miesiącach było istotnie większe w grupie pacjentek stosujących teryparatyd w porównaniu z grupą pacjentek przyjmujących alendronian i wynosiło: w przypadku lędźwiowej części kręgosłupa 4,2 % w porównaniu −1,9 %; p<0,001, dla kości biodra (3,8 % w porównaniu 0,9 %; p=0,005). Nie wykazano jednak istotnego wpływu na częstość złamań kości.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Dystrybucja Objętość dystrybucji wynosi około 1,7 l/kg mc. Okres półtrwania teryparatydu po podaniu podskórnym wynosi około 1 h i odpowiada czasowi absorpcji produktu leczniczego z miejsca wstrzyknięcia. Metabolizm Nie przeprowadzono badań dotyczących metabolizmu lub wydalania teryparatydu. Uważa się, że metabolizm obwodowy parathormonu zachodzi głównie w wątrobie i nerkach. Eliminacja Wydalanie teryparatydu zachodzi na drodze klirensu wątrobowego i pozawątrobowego (ok. 62 l/h u kobiet i 94 l/h u mężczyzn). Osoby w podeszłym wieku Nie stwierdzono różnic w farmakokinetyce teryparatydu w zależności od wieku (zakres wieku 31- 85 lat). Nie ma konieczności modyfikacji dawki w zależności od wieku pacjenta.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W standardowym zestawie testów nie stwierdzono genotoksycznych właściwości teryparatydu. Nie wykazywał także działania teratogennego w badaniach na szczurach, myszach i królikach. Nie obserwowano znaczącego wpływu u ciężarnych samic szczurów lub myszy, którym podawano teryparatyd w dawkach dobowych od 30 do 1 000 μg/kg mc. U ciężarnych samic królików, którym podawano teryparatyd w dawkach dobowych od 3 do 100 μg/kg mc. obserwowano resorpcję płodu i zmniejszenie liczebności miotu. Obserwowany u królików toksyczny wpływ na zarodek może wynikać z ich znacznie większej wrażliwości na wpływ parathormonu (PTH) na stężenie zjonizowanego wapnia we krwi w porównaniu z gryzoniami. U szczurów, którym prawie przez całe życie codziennie podawano teryparatyd we wstrzyknięciach, obserwowano proporcjonalny do stosowanych dawek nadmierny przyrost kości i zwiększoną częstość występowania kostniakomięsaka, prawdopodobnie w wyniku zmian aktywności genów.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclPrzedkliniczne dane o bezpieczeństwie
Teryparatyd nie powodował wzrostu częstości występowania innych nowotworów u szczurów. Znaczenie kliniczne tych danych jest prawdopodobnie niewielkie ze względu na różnice w fizjologii kości u ludzi i szczurów. U operacyjnie pozbawionych jajników małp, którym podawano produkt przez okres 18 miesięcy, nie stwierdzono przypadków guzów kości podczas leczenia ani przez kolejne 3 lata po jego zakończeniu. Ponadto w badaniach klinicznych ani w przeprowadzonym po ich zakończeniu badaniu obserwacyjnym nie odnotowano ani jednego przypadku kostniakomięsaka. W badaniach na zwierzętach wykazano, że znaczne ograniczenie przepływu krwi przez wątrobę zmniejsza kontakt PTH z głównym układem rozkładającym ten hormon (komórki Kupffera), a co za tym idzie klirens PTH(1-84).
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Kwas octowy lodowaty Mannitol Metakrezol Sodu octan bezwodny Rozcieńczony kwas solny (do ustalenia pH) Sodu wodorotlenek (do ustalenia pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 24 miesiącce. Wykazano trwałość chemiczną, fizyczną i mikrobiologiczną stosowanego produktu w okresie 28 dni w temperaturze 2-8°C. Po otwarciu produkt leczniczy można przechowywać nie dłużej niż 28 dni w temperaturze 2°C do 8°C. Bezpośrednio po każdym użyciu wstrzykiwacz należy ponownie umieścić w lodówce. Nie należy przechowywać wstrzykiwacza z założoną igłą. Nie należy wyjmować wkładu z wstrzykiwacza po pierwszym użyciu, Wkład we wstrzykiwaczu można dodatkowo umieścić w torebce dostarczanej ze wstrzykiwaczem w celu ochrony przed światłem.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
Za inne warunki i czas przechowywania stosowanego produktu odpowiedzialność ponosi użytkownik. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2°C – 8°C). Nie zamrażać. Wkład należy przechowywać w zewnętrznym opakowaniu w celu ochrony przed światłem. Warunki przechowywania po pierwszym otwarciu produktu leczniczego, patrz punkt 6.3 6.5 Rodzaj i zawartość opakowania 3 ml wkład (wkład ze szkła USP typu I), zamknięty korkiem (bromobutylowym) i, zatyczką (aluminium i gumowa uszczelka), umieszczony na plastikowych tackach pokrytych folią i dostarczany w tekturowych pudełkach. Jeden wkład zawiera 3 ml roztworu do wstrzykiwań, co odpowiada 28 dawkom po 20 mikrogramów każda (w 80 mikrolitrach). Wielkość opakowań: 1 wkład lub 3 wkłady. Nie wszystkie wielkości opakowań muszą się znajdować w obrocie.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
6.6 Specjalne środki ostrożności dotyczące usuwania Obsługa Wkłady leku Kauliv należy stosować wyłącznie z wstrzykiwaczem wielokrotnego użytku Kauliv Pen, będącym urządzeniem do podawania leków wielodawkowych. Do tego produktu leczniczego nie ma dołączonych igieł. Każdy wkład i wstrzykiwacz przeznaczony jest do stosowania wyłącznie przez jednego pacjenta. Wstrzykiwacz może być stosowany z igłami 32 G 4 mm Do każdego wstrzyknięcia musi być użyta nowa jałowa igła Termin ważności umieszczony na wkładzie musi być zawsze sprawdzany przed włożeniem wkładu do wstrzykiwacza Kauliv Pen. Upewnij się, że pozostało co najmniej 28 dni do upłynięcia daty ważności, żeby uniknąć pomyłek. Przed pierwszym użyciem wstrzykiwacza należy zapoznać się z informacjami o sposobie użycia wstrzykiwacza, zamieszczonymi w instrukcji użycia i je zrozumieć. Po każdym wstrzyknięciu, wstrzykiwacz należy ponownie umieścić w lodówce. Po pierwszym użyciu, nie należy wyjmować wkładu ze wstrzykiwacza przez 28 dni.
- CHPL leku Kauliv, roztwór do wstrzykiwań, 20 mcg/80 mclDane farmaceutyczne
Nie należy używać produktu Kauliv, jeśli jest lub był zamarznięty. Leku Kauliv nie wolno przenosić do strzykawki. Nie wolno ponownie napełniać pustego wkładu. Nie należy stosować produktu Kauliv, jeżeli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Data pierwszego wstrzyknięcia powinna być również zapisana na opakowaniu zewnętrznym wkładu Kauliv (patrz odpowiednie miejsce na pudełku: „Pierwsze użycie:”). Wstrzykiwacz wielorazowego użytku Kauliv ma pokrętło wyboru dawki, które za pomocą wyraźnych kliknięć i wskaźników wzrokowych zapewnia wybór właściwej dawki do napełniania wstępnego (P) oraz przy ustawianiu dawki (D) Usuwanie Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Produkt leczniczy Yorvipath zawiera PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez łącznik. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 6, 9 lub 12 mikrogramów PTH(1-34).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 15, 18 lub 21 mikrogramów PTH(1-34). Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 24, 27 lub 30 mikrogramów PTH(1-34). * Moc wskazuje ilość PTH(1-34) bez uwzględniania łącznika mPEG. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (płyn do wstrzykiwań) Przezroczysty i bezbarwny o pH 3,7–4,3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Yorvipath to terapia zastępcza parathormonem (PTH) wskazana do leczenia dorosłych z przewlekłą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
4.2 Dawkowanie i sposób podawania Leczenie powinno być rozpoczęte i monitorowane przez lekarzy lub wykwalifikowany personel medyczny doświadczony w diagnostyce oraz leczeniu pacjentów z niedoczynnością przytarczyc. Dawkowanie Zalecene dawki produktu leczniczego Yorvipath odnoszą się do mikrogramów PTH(1-34). Dawka powinna być dostosowana indywidualnie na podstawie stężenia wapnia w surowicy. Optymalna dawka po dostosowaniu to minimalna dawka wymagana do zapobiegania hipokalcemii. Jest to dawka, która utrzymuje stężenie wapnia w surowicy w prawidłowym zakresie bez potrzeby suplementacji czynnymi formami witaminy D lub wapniem w zakresie prawidłowym wykraczającym poza zalecaną suplementację żywieniową dla populacji ogólnej (ogólnie mniej niż 600 mg na dobę). Dawki czynnych postaci witaminy D i suplementów wapnia będą wymagać dostosowania przed rozpoczęciem i w trakcie leczenia produktem leczniczym Yorvipath na podstawie wartości wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Pacjenci otrzymujący maksymalną dawkę produktu leczniczego Yorvipath wynoszącą 60 µg na dobę, którzy doświadczają ciągłej hipokalcemii, mogą wymagać jednoczesnego podawania terapeutycznego wapnia i (lub) czynnych postaci witaminy D. Przed rozpoczęciem podawania produktu leczniczego Yorvipath Stężenie 25(OH) witaminy D w surowicy powinno być w zakresie prawidłowym, a stężenie wapnia w surowicy powinno być stabilne w zakresie prawidłowym lub nieco poniżej zakresu prawidłowego (1,95–2,64 mmol/l [7,8–10,6 mg/dl]) na co najmniej 1 wartość laboratoryjną na co najmniej dwa tygodnie przed pierwszą dawką leczenia. Rozpoczęcie podawania produktu leczniczego Yorvipath Zalecana dawka początkowa wynosi 18 µg raz na dobę ze stopniowym dostosowaniem dawki o 3 µg co 7 dni (patrz rysunek 1). Zakres dawki wynosi od 6 do 60 µg na dobę. Podczas rozpoczynania leczenia produktem leczniczym Yorvipath dawki czynnych postaci witaminy D lub suplementów wapnia powinny zostać dostosowane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
W razie przyjmowania czynnej witaminy D: o Jeżeli stężenie wapnia w surowicy wynosi ≥ 2,07 mmol/l [≥ 8,3 mg/dl/l], czynną witaminę D (kalcytriol lub alfakalcydol) należy odstawić tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. o Jeżeli stężenie wapnia w surowicy wynosi < 2,07 mmol/l [< 8,3 mg/dl/l], czynną witaminę D należy zmniejszyć o ≥ 50% tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. W razie nieprzyjmowania czynnej witaminy D: o Suplementy wapnia należy zmniejszyć o co najmniej 1500 mg tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. W razie przyjmowania dawek wapnia ≤ 1500 mg na dobę suplementy wapnia należy odstawić całkowicie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Jeżeli suplementy wapnia są wskazane w celu spełnienia wymogów dietetycznych, można rozważyć dalsze przyjmowanie suplementów wapnia w dawkach ≤ 600 mg na dobę zamiast ich całkowitego odstawienia. Dostosowanie i podtrzymywanie dawki produkt leczniczego Yorvipath Stężenie wapnia w surowicy musi być monitorowane w trakcie dostosowywania dawki (patrz punkt 4.4). Dawkę produktu leczniczego Yorvipath można stopniowo zwiększać o 3 µg, jeżeli upłynęło co najmniej 7 dni od wcześniejszej zmiany dawki (patrz rysunek 1). Nie wolno zwiększać dawki częściej niż co 7 dni. Dawkę produktu leczniczego Yorvipath można stopniowo zmniejszać o 3 µg nie częściej niż co 3 dni w odpowiedzi na hiperkalcemię (patrz rysunek 1). Stężenie wapnia w surowicy należy oznaczyć 7 dni po pierwszej dawce, oraz należy przestrzegać prawidłowego dawkowania produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementu wapnia zgodnie z zasadami przedstawionymi na rysunku 1.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Po każdej kolejnej zmianie dawki produktu leczniczego Yorvipath, czynnej witaminy D lub suplementów wapnia należy oznaczyć stężenie wapnia w surowicy w ciągu od 7 do 14 dni; pacjenci powinni być monitorowani pod kątem objawów klinicznych hipokalcemii lub hiperkalcemii. Dawki produktu leczniczego Yorvipath, czynnej witaminy D i (lub) suplementów wapnia należy dostosować zgodnie z rysunkiem 1. Dostosowywanie dawek produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia należy dokonywać tego samego dnia. Dawka podtrzymująca powinna być dawką, która zapewnia stężenie wapnia w surowicy w zakresie prawidłowym bez potrzeby suplementacji czynną witaminą D lub dawkami terapeutycznymi wapnia. Opcjonalnie można kontynuować suplementację wapniem wystarczającą do spełnienia wymogów dietetycznych (≤ 600 mg na dobę). Stężenie wapnia i 25(OH) witaminy D w surowicy należy oznaczać zgodnie ze standardem opieki po osiągnięciu dawki podtrzymującej.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Suplementacja 25(OH) witaminą D (nieczynną witaminą D) może być konieczna w celu osiągnięcia prawidłowych stężeń w surowicy. Rysunek 1: Dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia Małe stężenie wapnia w surowicy (< 2,07 mmol/l [< 8,3 mg/dl]): Tak ≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Kontynuować te same dawki suplementu wapnia i czynnej witaminy D na podstawie oceny klinicznej lekarza prowadzącego i zwiększyć tę samą dawkę produktu leczniczego Yorvipath o 3 µg Zwiększyć dawki suplementów wapnia i (lub) czynnej witaminy D do wcześniejszych dawek na podstawie oceny klinicznej lekarza prowadzącego i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Stężenie wapnia w surowicy w normie (od ≥ 2,07 do < 2,64 mmol/l [od ≥ 8,3 do < 10,6 mg/dl]): Nie Tak Dalsze przyjmowanie czynnej witaminy D?
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Kontynuować te same dawki produktu leczniczego Yorvipath, suplementu wapnia i czynnej witaminy D Zmniejszyć lub odstawić czynną witaminę D i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Tak Dalsze przyjmowanie suplementów wapnia? Kontynuować tę samą dawkę produktu leczniczego Yorvipath Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć suplementy wapnia o ≥ 1500 mg i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Odstawić suplementy wapnia i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Tak - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Nie Nie Nie Duże stężenie wapnia w surowicy (od ≥ 2,65 do < 3,00 mmol/l [od ≥ 10,7 do < 12,0 mg/dl]): Tak Dalsze przyjmowanie czynnej witaminy D? Dalsze przyjmowanie suplementów wapnia? Zmniejszyć lub odstawić czynną witaminę D i kontynuować te same dawki produktu leczniczego Yorvipath oraz suplementów wapnia Nie Nie Tak Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć dawkę produktu leczniczego Yorvipath o 3 µg Zmniejszyć suplementy wapnia o ≥ 1500 mg i kontynuować tę samą dawkę produktu leczniczego Yorvipath Odstawić suplementy wapnia i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Bardzo duże stężenie wapnia w surowicy (≥ 3,00 mmol/l [≥ 12 mg/dl]): Leczenie należy wstrzymać na 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Jeżeli kolejne stężenie wapnia w surowicy jest < 3,00 mmol/l [< 12 mg/dl], należy wznowić dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z odpowiednim punktem rysunku 1, stosując najnowszą uzyskaną wartość stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Jeżeli stężenie wapnia w surowicy pozostaje ≥ 3,00 mmol/l [≥ 12 mg/dl], należy wstrzymać podawanie produktu leczniczego Yorvipath na dodatkowe 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Więcej informacji na temat hiperkalcemii, patrz punkt 4.4. Pominięta dawka Jeśli pominięto dawkę o mniej niż 12 godzin, należy ją podać jak najszybciej. Jeśli dawkę pominięto o więcej niż 12 godzin, należy ją pominąć, a następną dawkę podać zgodnie z planem. Przerwanie lub odstawienie produktu leczniczego Yorvipath Należy unikać przerywania codziennego podawania w celu zminimalizowania wahań stężenia PTH w surowicy. Przerwanie lub odstawienie leczenia może spowodować hipokalcemię. Po przerywaniu lub odstawieniu leczenia na 3 lub więcej kolejnych dni należy obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii oraz rozważyć oznaczenie wapnia w surowicy. Jeżeli jest to wskazane, należy wznowić leczenie suplementami wapnia i czynną witaminą D.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Wznowić leczenie przepisaną dawką jak najszybciej po przerwaniu. Podczas wznawiania leczenia po przerwie oznaczyć stężenie wapnia w surowicy i dostosować dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z rysunkiem 1. Szczególne grupy pacjentów Osoby w starszym wieku Nie jest wymagane dostosowanie dawki zależnie od wieku (patrz punkt 5.2). Zaburzenie czynności wątroby Produkt leczniczy Yorvipath nie był badany u pacjentów z ciężkimi zaburzeniami czynności wątroby i u tych pacjentów powinien być stosowany z zachowaniem ostrożności (patrz punkt 4.4). Zaburzenie czynności nerek Nie jest wymagane dostosowanie dawki u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej (eGFR) ≥ 30 ml/min. W przypadku stosowania produktu leczniczego Yorvipath u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min zalecane jest częstsze oznaczanie stężenia wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Produkt leczniczy Yorvipath nie był badany u pacjentów z niedoczynnością przytarczyc oraz ciężkimi zaburzemiami czynności nerek (szacowany współczynnik filtracji kłębuszkowej < 30 ml/min) (patrz punkt 5.2). Dzieci i młodzież Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności produktu leczniczego Yorvipath u dzieci w wieku poniżej 18 lat. Dane nie są dostępne. Sposób podawania Produkt leczniczy Yorvipath musi być wstrzyknięty podskórnie w brzuch lub przednią część uda. Miejsca wstrzyknięcia należy codziennie zmieniać, wybierając jedno z czterech możliwych miejsc: brzuch (lewa lub prawa strona) oraz przednia część uda (lewego lub prawego). Dawki > 30 µg na dobę (kolejne wstrzyknięcia) Wszystkie dawki > 30 µg na dobę należy podawać jako dwie pojedyncze dawki wstrzykiwane kolejno w różne miejsca wstrzyknięć (tabela 1).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Zaleca się stosowanie innego wstrzykiwacza produktu leczniczego Yorvipath do drugiego wstrzyknięcia danego dnia, nawet jeżeli dwa wstrzykiwacze mają przycisk tego samego koloru (taka sama moc). Tabela 1: Zalecany schemat dawkowania produktu leczniczego Yorvipath > 30 µg/dobę
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
Dawka Schemat dawkowania Połączenie wstrzykiwaczy 33 µg/dobę 15 µg/dobę + 18 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)* 36 µg/dobę 18 µg/dobę + 18 µg/dobę 39 µg/dobę 18 µg/dobę + 21 µg/dobę 42 µg/dobę 21 µg/dobę + 21 µg/dobę 45 µg/dobę 21 µg/dobę + 24 µg/dobę Jeden fabrycznie napełniony wstrzykiwacz produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)+Jeden fabrycznie napełniony produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk)** 48 µg/dobę 24 µg/dobę + 24 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk) 51 µg/dobę 24 µg/dobę + 27 µg/dobę 54 µg/dobę 27 µg/dobę + 27 µg/dobę 57 µg/dobę 27 µg/dobę + 30 µg/dobę 60 µg/dobę 30 µg/dobę + 30 µg/dobę - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDawkowanie
* Produkt leczniczy Yorvipath 294 mikrogramy/0,98 ml dostarcza dawki 15, 18 lub 21 µg PTH(1-34) (z pomarańczowym przyciskiem) ** Produkt leczniczy Yorvipath 420 mikrogramów/1,4 ml dostarcza dawki 24, 27 lub 30 µg PTH(1-34) (z bordowym przyciskiem)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1; - Pacjenci z rzekomą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkich zdarzeń hiperkalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe podczas rozpoczynania leczenia lub zwiększania dawki. W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy (patrz punkt 4.2) i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hiperkalcemii. Jeżeli wystąpi ciężka hiperkalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath (patrz punkt 4.2). Hipokalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkiej hipokalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe po nagłym odstawieniu leczenia, ale może wystąpić w dowolnym momencie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlSpecjalne środki ostrozności
W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii. Jeżeli wystąpi ciężka hipokalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath i dostosować stałe lub doraźne dawki czynnej witaminy D i (lub) suplementów wapnia (patrz punkt 4.2). Stosowanie jednocześnie z glikozydami nasercowymi Hiperkalcemia z dowolnego powodu może predysponować do toksyczności naparstnicy. U pacjentów stosujących produkt leczniczy Yorvipath jednocześnie z glikozydami nasercowymi (takimi jak digoksyna lub digitoksyna) należy monitorować stężenia wapnia oraz glikozydu nasercowego w surowicy, a pacjentów obserwować pod kątem objawów podmiotowych i przedmiotowych toksyczności naparstnicy (patrz punkt 4.5).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlSpecjalne środki ostrozności
Ciężka choroba nerek lub wątroby Nie przeprowadzono badań z udziałem pacjentów z ciężkimi zaburzeniami czynności nerek i ciężkimi zaburzeniami czynności wątroby. W przypadku tych pacjentów produkt leczniczy Yorvipath powinien być stosowany z zachowaniem ostrożności. Pacjenci z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min mogą być bardziej podatni na hiperkalcemię oraz przemijające obniżenie szacowanego współczynnika filtracji kłębuszkowej, szczególnie na początku leczenia. Jeżeli leczenie jest rozpoczynane u takich pacjentów, zaleca się monitorowanie stężeń wapnia w surowicy. Stosowanie u pacjentów ze zwiększonym ryzkiem kostniakomięsaka Produkt leczniczy Yorvipath nie był badany i powinien być stosowany z zachowaniem ostrożności u pacjentów: - z nowotworami szkieletu oraz przerzutami w kościach; - którzy otrzymują lub otrzymywali radioterapię układu kostnego; - z niewyjaśnionym wzrostem poziomu aktywności fosfatazy alkalicznej swoistej dla kości; - z chorobami metabolicznymi kości, którzy są narażeni na podwyższone bazowe ryzyko kostniakomięsaka (np.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlSpecjalne środki ostrozności
choroba kości Pageta). Stosowanie u pacjentów z osteoporozą Badania przesiewowe i monitorowanie pod kątem osteoporozy powinno być zgodne z lokalnymi praktykami klinicznymi w przypadku każdego pacjenta ze zwiększonym ryzykiem złamań z powodu kruchości kości (patrz punkt 4.8). Zawartość sodu Lek zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że produkt leczniczy uznaje się za „wolny od sodu”.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji Nie przeprowadzono badań dotyczących interakcji z innymi produktami leczniczymi. Glikozydy nasercowe (takie jak digoksyna lub digitoksyna) mają wąski wskaźnik terapeutyczny, a na ich działanie wpływa wapń. U pacjentów przyjmujących produkt leczniczy Yorvipath oraz glikozydy nasercowe należy obserwować, czy nie występują objawy przedmiotowe i podmiotowe toksyczności naparstnicy. Inne produkty lecznicze mogące wywierać wpływ na stężenie wapnia w surowicy oraz zmieniać odpowiedź terapeutyczną produktu leczniczego Yorvipath to między innymi bisfosfoniany, denosumab, romosozumab, tiazyd oraz diuretyki pętlowe, kortykosteroidy układowe oraz lit. U pacjentów leczonych jednocześnie tymi produktami leczniczymi należy obserwować zmiany stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Ciąża Nie ma danych lub istnieją tylko ograniczone dane dotyczące stosowania produktu leczniczego Yorvipath u kobiet w ciąży. Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na reprodukcję (patrz punkt 5.3). Jednak nie można wykluczyć zagrożenia dla kobiet w ciąży lub rozwijającego się płodu. Decyzja o rozpoczęciu lub odstawieniu leczenia produktem leczniczym Yorvipath w trakcie ciąży powinna uwzględniać stosunek możliwych zagrożeń do korzyści dla kobiet w ciąży. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet w ciąży z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Karmienie piersią Nie wiadomo, czy palopegteryparatyd przenika do mleka ludzkiego. Ponieważ palopegteryparatyd nie jest wchłaniany po podaniu doustnym, jest mało prawdopodobne, aby miał niekorzystny wpływ na dziecko karmione piersią.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWpływ na płodność, ciążę i laktację
Podejmując decyzję o zaprzestaniu karmienia piersią lub leczenia produktem leczniczym Yorvipath, należy wziąć pod uwagę korzyści z karmienia piersią dla dziecka i korzyści z leczenia dla kobiety. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet karmiących piersią z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Płodność Nie przeprowadzono badań wpływu palopegteryparatydu na płodność człowieka. Dane z badań na zwierzętach nie wskazują, aby podawanie palopegteryparatydu upośledzało płodność (patrz punkt 5.3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Produkt leczniczy Yorvipath nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednak u niektórych pacjentów obserwowano zawroty głowy, stan przedomdleniowy, omdlenia i (lub) niedociśnienie ortostatyczne. Tacy pacjenci powinni powstrzymywać się od prowadzenia pojazdów i obsługiwania maszyn do czasu ustąpienia objawów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi w badaniach klinicznych z użyciem palopegteryparatydu były reakcje w miejscu wstrzyknięcia (21,6%), ból głowy (18,7%) i parestezja (13,7%). Najpoważniejszą reakcją niepożądaną zgłaszaną w badaniach klinicznych była hiperkalcemia (1,40%). Tabelaryczna lista reakcji niepożądanych Tabela 2 przedstawia reakcje niepożądane u pacjentów leczonych palopegteryparatydem zidentyfikowane we wszystkich badaniach fazy II oraz fazy III przeprowadzonych z grupami kontrolnymi otrzymującymi placebo. Reakcje niepożądane wymienione w poniższej tabeli przedstawiono według klasyfikacji układów i narządów MedDRA oraz częstości występowania zdefiniowanej za pomocą następującej konwencji: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000) i nieznana (częstość występowania nie może być określona na podstawie dostępnych danych).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
W obrębie każdej kategorii częstości występowania reakcje niepożądane przedstawiono według malejącej ciężkości. Tabela 2: Częstość występowania reakcji niepożądanych palopegteryparatydu
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
Klasyfikacja układówi narządów MedDRA Częstość występowania Reakcja niepożądana Zaburzenia metabolizmui odżywiania często hiperkalcemiaa, hipokalcemia Zaburzenia układu nerwowego bardzo często ból głowyd, parestezjaa często zawroty głowya, c, d, omdlenied,stan przedomdleniowyd Zaburzenia serca często palpitacjed, zespół posturalnej tachykardii ortostatycznejd Zaburzenia naczyniowe często niedociśnienie ortostatyczned niezbyt często nadciśnieniee Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia często ból jamy ustnej i gardła Zaburzenia żołądka i jelit bardzo często nudnościa często biegunkaa, zaparcia, wymioty,dyskomfort w jamie brzusznej, ból w jamie brzusznej Zaburzenia skóry i tkanki podskórnej często wysypka, reakcja nadwrażliwości na światło Zaburzenia mięśniowo- szkieletowe i tkanki łącznej często ból stawów, mialgia, drżenie mięśnif, ból mięśniowo-szkieletowyf Zaburzenia nerek i dróg moczowych niezbyt często nokturiae częstość nieznana wielomocze Zaburzenia ogólne i stany w miejscu podania bardzo często reakcje w miejscu wstrzyknięciaa,b, zmęczenie często astenia, pragnienie niezbyt często dyskomfort w klatce piersiowejf, ból w klatce piersiowejf Badania diagnostyczne częstość nieznana gęstość kości zmniejszona - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
a W przypadku tych reakcji niepożądanych pierwsze wystąpienie pojawiało się niemal wyłącznie w ciągu pierwszych 3 miesięcy leczenia (okres dostosowywania dawki). b Reakcje w miejscu wstrzyknięcia obejmują reakcję w miejscu wstrzyknięcia, rumień w miejscu wstrzyknięcia, zasiniaczenie w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia, krwotok w miejscu wstrzyknięcia, wysypkę w miejscu wstrzyknięcia oraz opuchliznę w miejscu wstrzyknięcia. c Zawroty głowy obejmują zawroty głowy i posturalne zawroty głowy. d Objawy związane z rozszerzeniem naczyń krwionośnych obejmowały posturalne zawroty głowy, ból głowy, palpitacje, zespół posturalnej tachykardii ortostatycznej, niedociśnienie ortostatyczne, obniżone ortostatyczne ciśnienie krwi i omdlenie. Objawy związane z rozszerzeniem naczyń krwionośnych (zidentyfikowane w badaniach klinicznych) występowały częściej podczas pierwszych 3 miesięcy leczenia i stanowiły podzbiór wszystkich zdarzeń zgłaszanych jako reakcje niepożądane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
W badaniu TCP-304 w ciągu pierwszych 3 miesięcy wystąpiły łącznie 3 zdarzenia (u 2 pacjentów) uznane za związane z palopegteryparatydem: posturalne zawroty głowy (n=1) oraz ból głowy i palpitacje (n=1). e Objawy przedmiotowe i podmiotowe potencjalnie związane z hiperkalcemią, jak zaobserwowano w badaniach klinicznych. f Objawy przedmiotowe i podmiotowe potencjalnie związane z hipokalcemią, jak zaobserwowano w badaniach klinicznych. Opis wybranych reakcji niepożądanych Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki hiperkalcemii. Częstość występowania hiperkalcemii była wyższa u pacjentów leczonych produktem leczniczym Yorvipath w porównaniu z placebo. W trakcie okresu zaślepionego objawową hiperkalcemię zgłoszono u 8,6% pacjentów leczonych produktem leczniczym Yorvipath i wszystkie zgłoszone przypadki wystąpiły w ciągu pierwszych 3 miesięcy po rozpoczęciu leczenia produktem leczniczym Yorvipath.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
Immunogenność U pacjentów mogą pojawić się przeciwciała przeciwko palopegteryparatydowi. Odsetek pacjentów z dodatnim wynikiem testu na przeciwciała wiążące był niski w dowolnym momencie leczenia — 0,7% miało niskie miano przeciwciał nieneutralizujących przeciwko PTH, a 5% miało niskie miano związanych z leczeniem przeciwciał przeciwko PEG. U 2,2% pacjentów leczonych palopegteryparatydem z wcześniej nabytymi przeciwciałami przeciwko PEG zaobserwowano przejściowy wpływ na PK (zwiększony klirens całkowitego PTH, mPEG i zmniejszenie stężenia PTH) oraz zmniejszenie stężenia wapnia w surowicy. Jednakże skuteczność terapeutyczna była utrzymana dzięki odpowiedniemu dostosowaniu dawki palopegteryparatydu zgodnie z algorytmem dostosowywania dawki w badaniu. Reakcje w miejscu wstrzyknięcia Reakcje w miejscu wstrzyknięcia były najczęściej zgłaszanymi w badaniach klinicznych reakcjami niepożądanymi (mediana czasu wystąpienia wynosiła 2,5 dnia; częstość występowania 21,6%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
Najczęstszymi reakcjami w miejscu wstrzyknięcia były rumień miejscowy (wszystkie o średnicy < 5 cm, a większość od 0 do < 2 cm) o nasileniu łagodnym lub umiarkowanym (stopień 1 lub 2) z medianą czasu trwania 72 godziny. Wszystkie reakcje w miejscu wstrzyknięcia ustępowały bez leczenia, żadna nie była ciężka ani nie prowadziła do przerwania leczenia. Objawy związane z rozszerzeniem naczyń krwionośnych Podczas stosowania leku Yorvipath zgłaszano objawy rozszerzenia naczyń krwionośnych. Objawy te są zwykle przemijające i ustępują bez leczenia; żaden nie był poważny ani nie doprowadził do przerwania leczenia. W przypadku wystąpienia objawów zaleca się dawkowanie przed snem, w pozycji leżącej. Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDziałania niepożądane
Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzedawkowanie
4.9 Przedawkowanie W razie przedawkowania pacjent powinien być obserwowany przez fachowy personel medyczny. Przedawkowanie może powodować hiperkalcemię, której objawy mogą obejmować odwodnienie, palpitacje serca, zmiany EKG, niedociśnienie, nudności, wymioty, zawroty głowy, osłabienie mięśni oraz splątanie. Pacjent z ciężką hiperkalcemią może wymagać obserwacji i opieki medycznej (patrz punkt 4.4). Jeden przypadek około 3-krotnego przypadkowego przedawkowania przepisanej dawki trwającego ponad 7 kolejnych dni spowodował stężenie wapnia w surowicy wynoszące aż 16,1 mg/dl, pacjent miał objawy i wymagał interwencji medycznej. Po wstrzymaniu podawania palopegteryparatydu, wapnia oraz czynnej witaminy D pacjent wyzdrowiał i ponownie rozpoczął przyjmowanie prawidłowej dawki.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: Hemostaza wapnia, parathormony i analogi, kod ATC: H05AA05 Mechanizm działania Endogenny parathormon (PTH) jest wydzielany przez przytarczyce jako polipeptyd o długości 84 aminokwasów. PTH wywiera działanie za pośrednictwem receptorów parathormonu na powierzchni komórek, na przykład ulegającego ekspresji w tkance kości, nerek i nerwów. Aktywacja PTH1R stymuluje obrót kostny, zwiększa wchłanianie zwrotne wapnia i wydalanie fosforanów w nerkach oraz ułatwia syntezę czynnej witaminy D. Palopegteryparatyd jest prolekiem zawierającym PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez zastrzeżony łącznik TransCon. PTH(1-34) i jego główny metabolit, PTH(1-33) mają podobne powinowactwo oraz aktywują PTH1R jak endogenny PTH. W warunkach fizjologicznych PTH jest w kontrolowany sposób odszczepiany od palopegteryparatydu zapewniając ciągłą ekspozycję ogólnoustrojową na aktywny PTH.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Skuteczność kliniczna i bezpieczeństwo stosowania Badanie z udziałem pacjentów z ustaloną niedoczynnością przytarczyc W kluczowym badaniu klinicznym fazy III PaTHway (TCP-304) oceniano skuteczność i bezpieczeństwo stosowania produktu leczniczego Yorvipath jako terapii zastępującej PTH u osób dorosłych z niedoczynnością przytarczyc. 26-tygodniowy, prowadzony metodą podwójnie ślepej próby, kontrolowany placebo okres badania klinicznego obejmował pacjentów randomizowanych (3:1) do grupy przyjmującej produkt leczniczy Yorvipath w dawce początkowej 18 mikrogramów/dobę lub placebo podawane jednocześnie z terapią konwencjonalną (suplement wapnia i czynna witamina D). Randomizacja była stratyfikowana według etiologii niedoczynności przytarczyc (tj. po zabiegu chirurgicznym wobec wszystkich innych przyczyn).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Badane leczenie (palopegteryparatyd lub placebo) i terapia konwencjonalna były następnie dostosowywane według algorytmu dawkowania opartego na stężeniach wapnia w surowicy skorygowanych o stężenie albumin. Średnia wieku pacjentów podczas rekrutacji wynosiła 49 lat (od 19 do 78 lat; 12% miało ≥ 65 lat) i większość pacjentów była kobietami (78%) oraz rasy białej (93%). Osiemdziesiąt pięć procent (85%) miało niedoczynność przytarczyc nabytą po operacji szyi. Spośród pacjentów z innymi etiologiami niedoczynności przytarczyc 7 (8,5%) pacjentów miało chorobę idiopatyczną, 2 miało autoimmunologiczny zespół niedoczynności wielogruczołowej typu 1 (APS-1), 1 miał hipokalcemię autosomalną dominującą typu 1 (ADH1, mutacja CaSR), 1 miał zespół DiGeorge i 1 miał niedoczynności przytarczyc, głuchotę czuciowo-nerwową oraz zespół dysplazji nerek (HDR) (mutacja GATA3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Przed randomizacją wszyscy pacjenci zostali poddani trwającemu około 4 tygodnie okresowi przesiewowemu, podczas którego suplementy wapnia oraz czynnej witaminy D dostosowano, aby osiągnąć stężenie wapnia w surowicy skorygowane o stężenie albumin wynoszące od 1,95 do 2,64 mmol/l (od 7,8 do 10,6 mg/dl), stężenie magnezu ≥ 0,53 mmol/l (≥ 1,3 mg/dl) i poniżej górnego zakresu referencyjnego normy oraz stężenie 25(OH) witaminy D od 50 do 200 nmol/l (od 20 do 80 ng/ml). W przypadku terapii konwencjonalnej pacjenci byli leczeni średnim wyjściowymi dawkami wapnia (pierwiastkowego) wynoszącymi 1839 mg/dobę. Średnie wyjściowe dawki czynnej witaminy D wynosiły 0,75 mikrograma/dobę u pacjentów leczonych kalcytriolem (n=70) oraz 2,3 mikrograma/dobę u pacjentów leczonych alfakalcydolem (n=12). Średnie wyjściowe stężenie wapnia w surowicy skorygowane o stężenie albumin oraz średnie 24-godzinne stężenie wapnia w moczu były podobne w obu grupach leczenia: średnie stężenie wapnia w surowicy wynosiło 2,2 mmol/l (8,8 mg/dl) oraz 2,15 mmol/l (8,6 mg/dl), a średnie 24-godzinne stężenie wapnia w moczu wynosiło 392 mg/dobę i 329 mg/dobę odpowiednio w przypadku produktu leczniczego Yorvipath oraz placebo.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Pierwszorzędowy punkt końcowy Pierwszorzędowy złożony punkt końcowy był definiowany jako odsetek pacjentów w tygodniu 26, którzy osiągnęli: stężenia wapnia w surowicy w zakresie prawidłowym (od 2,07 do 2,64 mmol/l [od 8,3 do 10,6 mg/dl]), niezależnie od terapii konwencjonalnej definiowanej jako niewymagająca czynnej witaminy D oraz ≤ 600 mg/dobę suplementacji wapnia i bez zwiększenia przepisanego badanego leczenia w ciągu 4 tygodni przed tygodniem 26. Kluczowe drugorzędowe punkty końcowe obejmowały podzbiór punktacji domeny skali doświadczeń pacjentów z niedoczynnością przytarczyc (ang. Hypoparathyroidism Patient Experience Scale, HPES) oraz punktacji podskali skróconego formularza 36-elementowego (ang. 36-Item Short Form Survey, SF-36). Liczbę pacjentów spełniających pierwszorzędowy złożony punkt końcowy w porównaniu z grupą placebo i każdym składnikiem pierwszorzędowego punktu końcowego w tygodniu 26 przedstawiono w tabeli 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Tabela 3: TCP-304: współczynnik odpowiedzi oparty na pierwszorzędowym punkcie końcowym w tygodniu 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Yorvipath (N=61) (n, %) Placebo (N=21) (n, %) Różnica współczynnikaodpowiedzi (95% CI) Odpowiedź w tygodniu 26 48 (78,7%) 1 (4,8%) 74,0%(60,4%; 87,6%)p < 0,0001 Odpowiedź dla każdego składnika Stężenie wapnia w surowicy skorygowaneo stężenie albumin w zakresie prawidłowyma 49 (80,3%) 10 (47,6%) 32,7%(9,2%; 56,3%) Niezależność od czynnej witaminy Db 60 (98,4%) 5 (23,8%) 74,6%(56,1%; 93,1%) Niezależność od terapeutycznych dawek wapniac 57 (93,4%) 1 (4,8%) 88,7%(77,7%; 99,7%) Brak zwiększenia dawki w grupie produktu leczniczego Yorvipathd 57 (93,4%) 12 (57,1%) 36,4%(14,2%; 58,5%) - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
a Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosił od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl). b Wszystkie codzienne stałe dawki czynnej witaminy D równe zero ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. c Średnie codzienne stałe dawki pierwiastkowego wapnia ≤ 600 mg ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. d Brak zwiększenia dawki produktu leczniczego Yorvipath w ciągu 4 tygodni przed wizytą po 26 tygodniach. Skróty: CI: przedział ufności; PRN: pro re nata. Drugorzędowe punkty końcowe Przyjmowanie terapii konwencjonalnej: dawki wapnia i czynnej witaminy D. W badaniu fazy III PaTHway, w tygodniu 26, 93% (57/61) pacjentów w grupie produktu leczniczego Yorvipath było w stanie odstawić terapię konwencjonalną (tj. odstawić czynną witaminę D oraz terapeutyczne dawki wapnia).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Wszyscy pacjenci w grupie produktu leczniczego Yorvipath odstawili czynną witaminę D do tygodnia 8 i utrzymywali zmniejszenie terapeutycznych dawek wapnia. Występowało znaczące zmniejszenie przyjmowania terapii konwencjonalnej w grupie produktu leczniczego Yorvipath od wartości wyjściowej do tygodnia 26 w porównaniu z placebo: czynnej witaminy D (nominalna wartość p < 0,0001), dawki wapnia (nominalna wartość p = 0,0003) oraz codziennego obciążenia tabletkami (nominalna wartość p < 0,0001) (tabela 4). Tabela 4: Drugorzędowe punkty końcowe: przyjmowanie terapii konwencjonalnej w tygodniu 26 — okres zaślepiony (populacja ITT)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Yorvipath (n/N=60/61)a Placebo (n/N=19/21)a Nominalnawartość p Wartość początkowa Tydzień 26 Wartość początkowa Tydzień 26 Suplementacyjna dawkaczynnej witaminy D (µg), średnia (SD) 1,0 (0,7) 0,0 (0,0) 1,0 (0,6) 0,6 (0,7) < 0,0001 Suplementacyjna dawka wapnia (mg), średnia (SD) 1737 (907) 274 (177) 2089 (1448) 1847 (1326) 0,0003 Codzienne obciążenie tabletkami (liczba tabletekterapii konwencjonalnej), średnia (SD) 6,6 (2,1) 0,5 (1,7) 6,3 (2,8) 5,4 (3,2) < 0,0001 - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Nominalna wartość p z badań różnic zmiany od wartości wyjściowej do tygodnia 26 między produktem leczniczym Yorvipath oraz placebo. a N to liczba pacjentów w populacji ITT; n to liczba pacjentów z danymi zarówno wyjściowymi, jak i w tygodniu 26. Parametry biochemiczne surowicy Średnia wartość wapnia w surowicy początkowo wzrosła i utrzymywała się w zakresie prawidłowym u pacjentów leczonych palopegteryparatydem (rysunek 2). U pacjentów przyjmujących placebo stężenia wapnia w surowicy nieco zmniejszyły się, spadając poniżej zakresu prawidłowego w tygodniu 2 (średnia obserwowana wartość: 2,06 mmol/l) i w tygodniu 26 (średnia obserwowana wartość: 2,06 mmol/l). Różnica średniej najmniejszych kwadratów leczenia między produktem leczniczym Yorvipath i placebo wynosiła 0,17 mmol/l (95% CI: 0,100; 0,247; nominalna wartość p < 0,0001) w tygodniu 26. Rysunek 2: Wapń w surowicy (średnia ±SE) według wizyty — okres zaślepiony (populacja ITT) 3 2,5 2 1,5 0 2 4 6 8 10 12 14 16 18 20 22 24 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Średnie (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mmol/l) Czas (tygodnie)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Palopegteryparatyd
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Placebo Średnie stężenia fosforanu w surowicy u pacjentów leczonych palopegteryparatydem były w zakresie prawidłowym wyjściowo i utrzymywały się w zakresie prawidłowym do tygodnia 26 (średnia zmiana od wartości wyjściowej do tygodnia 26 wynosiła -0,13 mmol/l). Średni iloczyn wapnia i fosforanów w surowicy zmniejszył się u pacjentów leczonych produktem leczniczym Yorvipath i pozostawał stabilny w zakresie prawidłowym do tygodnia 26. 24-godzinne wydalanie wapnia w moczu Terapia produktem leczniczym Yorvipath normalizowała 24-godzinną średnią wydalania wapnia w moczu i wykazywała większe zmniejszenie 24-godzinnego stężenia wapnia w moczu niż placebo. Dzieci i młodzież Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego Yorvipath w jednej lub kilku podgrupach populacji dzieci i młodzieży z niedoczynnością przytarczyc, zgodnie z warunkami zawartymi w decyzji dotyczącej planu badań dzieci i młodzieży (PIP, ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakodynamiczne
Paediatric Investigation Plan) we wskazaniu leczenia niedoczynności przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Wchłanianie Po codziennym podawaniu podskórnym palopegteryparatyd uwalnia PTH za pośrednictwem autoodszczepienia łącznika TransCon podlegając kinetyce pierwszorzędowej, dając ciągłą ekspozycję przez 24 godziny w szacowanym zakresie prawidłowym (rysunek 3). Rysunek 3: Średni uwalniany PTH* po podawaniu podskórnym palopegteryparatydu w stanie stacjonalnym u pacjentów z niedoczynnością przytarczyc 40 30 20 10 0 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
Średni (+/-SE) uwalniany PTH (pg/ml) Czas (godz.) Szacowany zakres prawidłowy dla PTH(1-34) wynosi około 4 do 26 pg/ml. Jest on obliczony na podstawie PTH(1-34) stanowiącego 40% masy cząsteczkowej PTH(1-84)** i zakresu prawidłowego (10 do 65 pg/ml) dla PTH(1-84). * Średnia dawka palopegteryparatydu (zakres): 22,3 (12-33) µg/dobę, n=7. uwolniony PTH: suma PTH(1-34) i PTH(1-33). ** PTH(1-84) = endogenny parathormon. U pacjentów z niedoczynnością przytarczyc podawanie palopegteryparatydu odpowiadało 18 µg PTH(1-34)/dobę, przewidywane maksymalne stężenie w osoczu (C max ) (CV%) palopegteryparatydu wynosiło 5,18 ng/ml (36%), a przewidywana wartość C max (CV%) dla uwalnianego PTH wynosiła 6,9 pg/ml (22%) z medianą czasu do osiągnięcia maksymalnych stężeń (T max ) wynoszącą 4 godziny. Przewidywana ekspozycja w ciągu 24-godzinnego odstępu między dawkami (pole pod krzywą, ang. area under the curve, AUC) (CV%) dla uwalnianego PTH wynosiła 150 pg*h/ml (22%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
Po podaniu wielu podskórnych dawek palopegteryparatydu w zakresie od 12 do 24 µg PTH(1-34)/dobę stężenia palopegteryparatydu i uwalnianego PTH rosły w sposób proporcjonalny do dawki, osiągając stan stacjonarny w ciągu odpowiednio około 10 i 7 dni. Stosunek wartości maksymalnej do minimalnej był niski, wynosił około 1,1 oraz 1,5 przez 24 godziny stanu stacjonarnego odpowiednio dla palopegteryparatydu i uwalnianego PTH. Palopegteryparatyd ulegał akumulacji po wielu dawkach do 18-krotności wartości AUC. Dystrybucja Pozorną objętość dystrybucji (CV%) palopegteryparatydu szacuje się na 4,8 l (50%) oraz do 8,7 l (18%) dla uwalnianego PTH. Metabolizm PTH uwalniany z palopegteryparatydu składa się z PTH(1-34) i metabolitu PTH(1-33). PTH jest metabolizowany i usuwany w nerkach. Eliminacja U zdrowych osób dorosłych klirens (CV%) palopegteryparatydu w stanie stacjonarnym szacuje się na 0,58 l/dobę (52%) z przewidywanym okresem półtrwania wynoszącym 70 godzin.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
Pozorny okres półtrwania PTH uwalnianego z palopegteryparatydu wynosi około 60 godzin. W wątrobie większość PTH jest rozszczepiana przez katepsyny. W nerkach niewielka ilość PTH wiąże się z PTH1R, ale większość jest wydalana poprzez filtrację kłębuszkową. Zależności farmakokinetyczno-farmakodynamiczne W cząstkowym badaniu farmakodynamiki/farmakokinetyki u pacjentów z niedoczynnością przytarczyc, codzienne podskórne podawanie palopegteryparatydu (średnia dawka (zakres): 22,3 (12– 33) µg PTH (1-34)dobę) zwiększała stężenie wapnia w surowicy do zakresu prawidłowego (patrz rysunek 4). Zwiększenie stężeń wapnia w surowicy następowało w sposób zależny od dawki wspierając możliwość dostosowania dawki palopegteryparatydu odpowiednio do zmierzonych wartości wapnia w surowicy u danego pacjenta. mmol/l Rysunek 4: Średnie stężenia wapnia w surowicy skorygowane o stężenie albumin po podawaniu podskórnym palopegteryparatydu w stanie stacjonarnym u pacjentów z niedoczynnością przytarczyc 12 11 10 9 8 7 3 2,75 2,5 2,25 2 1,75 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
Średnia (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mg/dl) Czas (godz.) Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosi od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl), co oznaczono szarym cieniowaniem. Średnia dawka palopegteryparatydu (zakres): 22,3 (12–33) µg PTH(1- 34)/dobę, n=7. Szczególne grupy pacjentów Na farmakokinetykę uwalnianego PTH nie wpływa płeć ani masa ciała. Dane dotyczące rasy i pochodzenia etnicznego nie wykazują żadnych trendów wskazujących na różnice, ale dostępne dane są zbyt ograniczone do postawienia rozstrzygających wniosków. Osoby w starszym wieku Na farmakokinetykę uwalnianego PTH nie wpływa wiek (od 19 do 76 lat). Zaburzenia czynności nerek Produkt leczniczy Yorvipath podawano pacjentom z niedoczynnością przytarczyc z eGFR ≥ 30 ml/min w długotrwałych badaniach klinicznych bez konieczności dostosowywania dawki poza algorytmem dostosowywania dawki w ramach badania.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlWłaściwości farmakokinetyczne
Nie przeprowadzono badań klinicznych z udziałem pacjentów z niedoczynnością przytarczyc oraz zaburzeniem czynności nerek (< 30 ml/min) lub dializowanych. W badaniu, w którym produkt leczniczy Yorvipath podawano jako pojedynczą dawkę uczestnikom bez niedoczynności przytarczyc, ekspozycja na palopegteryparatyd i uzyskiwane stężenia wapnia w surowicy były podobne jak u uczestników z łagodnym, umiarkowanym lub ciężkim upośledzeniem czynności nerek w porównaniu do pacjentów bez zaburzeń czynności nerek.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie Nie ujawniono szczególnego zagrożenia dla człowieka wynikającego z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, genotoksyczności i tolerancji miejscowej przeprowadzonych na palopegteryparatydzie. U zwierząt wszystkich zastosowanych gatunków podanie wielokrotne największych dawek powodowało niepożądaną uporczywą hiperkalcemię, która w niektórych badaniach prowadziła do przedwczesnego zgonu/eutanazji, objawów klinicznych, utrata masy ciała i (lub) mineralizacji tkanki miękkiej obserwowanej głównie w nerkach. Wyniki te są uważane za skutki nieustannie przesadnej farmakologii PTH i nie mają one znaczenia w warunkach klinicznych, gdzie dawkę dostosowuje się w celu zapewnienia normalizacji stężenia wapnia w surowicy Zgodnie z oczekiwanymi skutkami farmakologicznymi, powtarzane codzienne podawanie palopegteryparatydu zwiększało obrót kostny u szczurów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzedkliniczne dane o bezpieczeństwie
Przy małych dawkach (2-krotność maksymalnej zalecanej dawki dla człowieka, MRHD) na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny indukował netto całkowite kataboliczne skutki kostne. Przy dużych dawkach (5-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny powodował netto anaboliczny skutek kostny. Dysplazja nasad kości była obserwowana przy najwyższej dawce (9-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów. Skutki te nie mają znaczenia w warunkach klinicznych, w których dawki leku Yorvipath dostosowuje się indywidualnie. Nie było skutków sercowo-naczyniowych u małp do najwyższej badanej dawki (włącznie) w badaniach dawki pojedynczej (3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ) lub dawki powtarzanej (0,98-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzedkliniczne dane o bezpieczeństwie
Nasilone występowanie kostniakomięsaków obserwowano w badaniach rakotwórczości z krótko żyjącymi analogami PTH u szczurów, ale bez dowodów zwiększonego ryzyka kostniakomięsaka u pacjentów leczonych krótko żyjącymi analogami PTH.Dlatego też nie przeprowadzono żadnego badania nad rakotwórczością palopegteryparatydu. W badaniach reprodukcji na zwierzętach podawanie palopegteryparatydu ciężarnym szczurom i królikom w okresie organogenezy nie dawało dowodów na letalność zarodka, toksyczność dla płodu lub dysmorfogenezę w przypadku podania najwyższych badanych dawek (włącznie) (odpowiednio 8- i 7-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC). Skutki przesadnej farmakologii PTH obserwowano przy najwyższych badanych dawkach u ciężarnych szczurów oraz królików (podwyższone stężenia wapnia w surowicy, zmniejszona masa ciała, zmniejszone spożycie pokarmu i (lub) oznaki kliniczne). Ekspozycje przy poziomie dawkowania, przy którym nie obserwuje się szkodliwych zmian (ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlPrzedkliniczne dane o bezpieczeństwie
no observed adverse effect level, NOAEL) w przypadku toksyczności dla matki wynosił 2- i 3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC u odpowiednio ciężarnych szczurów i królików. Nie przeprowadzono badania rozwoju pre- i postnatalnego związanego z palopegteryparatydem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Kwas bursztynowy Mannitol Metakrezol Sodu wodorotlenek Kwas chlorowodorowy (do regulacji pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 3 lata. Po pierwszym otwarciu Przechowywać w temperaturze poniżej 30 °C. Pozostawić zatyczkę wstrzykiwacza na fabrycznie napełnionym wstrzykiwaczu w celu ochrony przed światłem. Produkt leczniczy Yorvipath należy wyrzucić po 14 dniach. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2 °C – 8 °C). Nie zamrażać. Przechowywać w oryginalnym opakowaniu z założoną zatyczką wstrzykiwacza w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po pierwszym otwarciu, patrz punkt 6.3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDane farmaceutyczne
6.5 Rodzaj i zawartość opakowania Wkład (szkło typu 1) z tłokiem (halobutyl) i laminowanym arkuszem gumowym (halobutyl/izopren) umieszczony w wielodawkowym jednorazowym fabrycznie napełnionym wstrzykiwaczu wykonanym z polipropylenu. Opakowania dwóch fabrycznie napełnionych wstrzykiwaczy oraz 30 igieł jednorazowych na 28 dni leczenia (zapakowane razem w wewnętrznych pudełkach). Każde wewnętrzne pudełko zawiera jeden fabrycznie napełniony wstrzykiwacz oraz 15 igieł na 14 dni leczenia. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 6, 9 lub 12 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest niebieski.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDane farmaceutyczne
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 15, 18 lub 21 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest pomarańczowy. Yorvipath 420 mikrogramy/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 24, 27 lub 30 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest bordowy. 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Przygotowanie dawki Nowy wstrzykiwacz produktu leczniczego Yorvipath należy wyjąć z lodówki na 20 minut przed pierwszym otwarciem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDane farmaceutyczne
Roztwór powinien być przezroczysty, bezbarwny i wolny od cząstek stałych. Nie należy wstrzykiwać produktu leczniczego, jeżeli jest mętny lub zawiera cząstki stałe. Każdy fabrycznie napełniony wstrzykiwacz jest przeznaczony do użytku przez jednego pacjenta. Fabrycznie napełniony wstrzykiwacz nie powinien być współdzielony przez pacjentów, nawet jeżeli igła jest zmieniana. Jeżeli fabrycznie napełniony wstrzykiwacz był zamrażany lub narażony na ciepło, należy go wyrzucić. Za każdym razem, gdy fabrycznie napełniony wstrzykiwacz jest przygotowywany do podawania, należy założyć nową igłę. Igieł nie wolno używać ponownie. Może to zapobiec zablokowaniu igieł, zanieczyszczeniu, zakażeniu, wyciekowi roztworu lub niedokładnemu dawkowaniu. Igła iniekcyjna powinna być zdejmowana po każdym wstrzyknięciu, a wstrzykiwacz należy przechowywać bez przymocowanej igły. Igłę należy wyrzucać po każdym wstrzyknięciu.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 168 mcg/0,56 mlDane farmaceutyczne
Instrukcje przygotowywania i podawania produktu leczniczego Yorvipath podano w ulotce dołączonej do opakowania oraz instrukcji użycia. Utylizacja Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Produkt leczniczy Yorvipath zawiera PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez łącznik. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 6, 9 lub 12 mikrogramów PTH(1-34).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 15, 18 lub 21 mikrogramów PTH(1-34). Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 24, 27 lub 30 mikrogramów PTH(1-34). * Moc wskazuje ilość PTH(1-34) bez uwzględniania łącznika mPEG. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (płyn do wstrzykiwań) Przezroczysty i bezbarwny o pH 3,7–4,3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Yorvipath to terapia zastępcza parathormonem (PTH) wskazana do leczenia dorosłych z przewlekłą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
4.2 Dawkowanie i sposób podawania Leczenie powinno być rozpoczęte i monitorowane przez lekarzy lub wykwalifikowany personel medyczny doświadczony w diagnostyce oraz leczeniu pacjentów z niedoczynnością przytarczyc. Dawkowanie Zalecene dawki produktu leczniczego Yorvipath odnoszą się do mikrogramów PTH(1-34). Dawka powinna być dostosowana indywidualnie na podstawie stężenia wapnia w surowicy. Optymalna dawka po dostosowaniu to minimalna dawka wymagana do zapobiegania hipokalcemii. Jest to dawka, która utrzymuje stężenie wapnia w surowicy w prawidłowym zakresie bez potrzeby suplementacji czynnymi formami witaminy D lub wapniem w zakresie prawidłowym wykraczającym poza zalecaną suplementację żywieniową dla populacji ogólnej (ogólnie mniej niż 600 mg na dobę). Dawki czynnych postaci witaminy D i suplementów wapnia będą wymagać dostosowania przed rozpoczęciem i w trakcie leczenia produktem leczniczym Yorvipath na podstawie wartości wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Pacjenci otrzymujący maksymalną dawkę produktu leczniczego Yorvipath wynoszącą 60 µg na dobę, którzy doświadczają ciągłej hipokalcemii, mogą wymagać jednoczesnego podawania terapeutycznego wapnia i (lub) czynnych postaci witaminy D. Przed rozpoczęciem podawania produktu leczniczego Yorvipath Stężenie 25(OH) witaminy D w surowicy powinno być w zakresie prawidłowym, a stężenie wapnia w surowicy powinno być stabilne w zakresie prawidłowym lub nieco poniżej zakresu prawidłowego (1,95–2,64 mmol/l [7,8–10,6 mg/dl]) na co najmniej 1 wartość laboratoryjną na co najmniej dwa tygodnie przed pierwszą dawką leczenia. Rozpoczęcie podawania produktu leczniczego Yorvipath Zalecana dawka początkowa wynosi 18 µg raz na dobę ze stopniowym dostosowaniem dawki o 3 µg co 7 dni (patrz rysunek 1). Zakres dawki wynosi od 6 do 60 µg na dobę. Podczas rozpoczynania leczenia produktem leczniczym Yorvipath dawki czynnych postaci witaminy D lub suplementów wapnia powinny zostać dostosowane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
W razie przyjmowania czynnej witaminy D: o Jeżeli stężenie wapnia w surowicy wynosi ≥ 2,07 mmol/l [≥ 8,3 mg/dl/l], czynną witaminę D (kalcytriol lub alfakalcydol) należy odstawić tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. o Jeżeli stężenie wapnia w surowicy wynosi < 2,07 mmol/l [< 8,3 mg/dl/l], czynną witaminę D należy zmniejszyć o ≥ 50% tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. W razie nieprzyjmowania czynnej witaminy D: o Suplementy wapnia należy zmniejszyć o co najmniej 1500 mg tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. W razie przyjmowania dawek wapnia ≤ 1500 mg na dobę suplementy wapnia należy odstawić całkowicie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Jeżeli suplementy wapnia są wskazane w celu spełnienia wymogów dietetycznych, można rozważyć dalsze przyjmowanie suplementów wapnia w dawkach ≤ 600 mg na dobę zamiast ich całkowitego odstawienia. Dostosowanie i podtrzymywanie dawki produkt leczniczego Yorvipath Stężenie wapnia w surowicy musi być monitorowane w trakcie dostosowywania dawki (patrz punkt 4.4). Dawkę produktu leczniczego Yorvipath można stopniowo zwiększać o 3 µg, jeżeli upłynęło co najmniej 7 dni od wcześniejszej zmiany dawki (patrz rysunek 1). Nie wolno zwiększać dawki częściej niż co 7 dni. Dawkę produktu leczniczego Yorvipath można stopniowo zmniejszać o 3 µg nie częściej niż co 3 dni w odpowiedzi na hiperkalcemię (patrz rysunek 1). Stężenie wapnia w surowicy należy oznaczyć 7 dni po pierwszej dawce, oraz należy przestrzegać prawidłowego dawkowania produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementu wapnia zgodnie z zasadami przedstawionymi na rysunku 1.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Po każdej kolejnej zmianie dawki produktu leczniczego Yorvipath, czynnej witaminy D lub suplementów wapnia należy oznaczyć stężenie wapnia w surowicy w ciągu od 7 do 14 dni; pacjenci powinni być monitorowani pod kątem objawów klinicznych hipokalcemii lub hiperkalcemii. Dawki produktu leczniczego Yorvipath, czynnej witaminy D i (lub) suplementów wapnia należy dostosować zgodnie z rysunkiem 1. Dostosowywanie dawek produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia należy dokonywać tego samego dnia. Dawka podtrzymująca powinna być dawką, która zapewnia stężenie wapnia w surowicy w zakresie prawidłowym bez potrzeby suplementacji czynną witaminą D lub dawkami terapeutycznymi wapnia. Opcjonalnie można kontynuować suplementację wapniem wystarczającą do spełnienia wymogów dietetycznych (≤ 600 mg na dobę). Stężenie wapnia i 25(OH) witaminy D w surowicy należy oznaczać zgodnie ze standardem opieki po osiągnięciu dawki podtrzymującej.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Suplementacja 25(OH) witaminą D (nieczynną witaminą D) może być konieczna w celu osiągnięcia prawidłowych stężeń w surowicy. Rysunek 1: Dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia Małe stężenie wapnia w surowicy (< 2,07 mmol/l [< 8,3 mg/dl]): Tak ≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Kontynuować te same dawki suplementu wapnia i czynnej witaminy D na podstawie oceny klinicznej lekarza prowadzącego i zwiększyć tę samą dawkę produktu leczniczego Yorvipath o 3 µg Zwiększyć dawki suplementów wapnia i (lub) czynnej witaminy D do wcześniejszych dawek na podstawie oceny klinicznej lekarza prowadzącego i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Stężenie wapnia w surowicy w normie (od ≥ 2,07 do < 2,64 mmol/l [od ≥ 8,3 do < 10,6 mg/dl]): Nie Tak Dalsze przyjmowanie czynnej witaminy D?
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Kontynuować te same dawki produktu leczniczego Yorvipath, suplementu wapnia i czynnej witaminy D Zmniejszyć lub odstawić czynną witaminę D i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Tak Dalsze przyjmowanie suplementów wapnia? Kontynuować tę samą dawkę produktu leczniczego Yorvipath Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć suplementy wapnia o ≥ 1500 mg i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Odstawić suplementy wapnia i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Tak - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Nie Nie Nie Duże stężenie wapnia w surowicy (od ≥ 2,65 do < 3,00 mmol/l [od ≥ 10,7 do < 12,0 mg/dl]): Tak Dalsze przyjmowanie czynnej witaminy D? Dalsze przyjmowanie suplementów wapnia? Zmniejszyć lub odstawić czynną witaminę D i kontynuować te same dawki produktu leczniczego Yorvipath oraz suplementów wapnia Nie Nie Tak Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć dawkę produktu leczniczego Yorvipath o 3 µg Zmniejszyć suplementy wapnia o ≥ 1500 mg i kontynuować tę samą dawkę produktu leczniczego Yorvipath Odstawić suplementy wapnia i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Bardzo duże stężenie wapnia w surowicy (≥ 3,00 mmol/l [≥ 12 mg/dl]): Leczenie należy wstrzymać na 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Jeżeli kolejne stężenie wapnia w surowicy jest < 3,00 mmol/l [< 12 mg/dl], należy wznowić dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z odpowiednim punktem rysunku 1, stosując najnowszą uzyskaną wartość stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Jeżeli stężenie wapnia w surowicy pozostaje ≥ 3,00 mmol/l [≥ 12 mg/dl], należy wstrzymać podawanie produktu leczniczego Yorvipath na dodatkowe 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Więcej informacji na temat hiperkalcemii, patrz punkt 4.4. Pominięta dawka Jeśli pominięto dawkę o mniej niż 12 godzin, należy ją podać jak najszybciej. Jeśli dawkę pominięto o więcej niż 12 godzin, należy ją pominąć, a następną dawkę podać zgodnie z planem. Przerwanie lub odstawienie produktu leczniczego Yorvipath Należy unikać przerywania codziennego podawania w celu zminimalizowania wahań stężenia PTH w surowicy. Przerwanie lub odstawienie leczenia może spowodować hipokalcemię. Po przerywaniu lub odstawieniu leczenia na 3 lub więcej kolejnych dni należy obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii oraz rozważyć oznaczenie wapnia w surowicy. Jeżeli jest to wskazane, należy wznowić leczenie suplementami wapnia i czynną witaminą D.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Wznowić leczenie przepisaną dawką jak najszybciej po przerwaniu. Podczas wznawiania leczenia po przerwie oznaczyć stężenie wapnia w surowicy i dostosować dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z rysunkiem 1. Szczególne grupy pacjentów Osoby w starszym wieku Nie jest wymagane dostosowanie dawki zależnie od wieku (patrz punkt 5.2). Zaburzenie czynności wątroby Produkt leczniczy Yorvipath nie był badany u pacjentów z ciężkimi zaburzeniami czynności wątroby i u tych pacjentów powinien być stosowany z zachowaniem ostrożności (patrz punkt 4.4). Zaburzenie czynności nerek Nie jest wymagane dostosowanie dawki u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej (eGFR) ≥ 30 ml/min. W przypadku stosowania produktu leczniczego Yorvipath u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min zalecane jest częstsze oznaczanie stężenia wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Produkt leczniczy Yorvipath nie był badany u pacjentów z niedoczynnością przytarczyc oraz ciężkimi zaburzemiami czynności nerek (szacowany współczynnik filtracji kłębuszkowej < 30 ml/min) (patrz punkt 5.2). Dzieci i młodzież Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności produktu leczniczego Yorvipath u dzieci w wieku poniżej 18 lat. Dane nie są dostępne. Sposób podawania Produkt leczniczy Yorvipath musi być wstrzyknięty podskórnie w brzuch lub przednią część uda. Miejsca wstrzyknięcia należy codziennie zmieniać, wybierając jedno z czterech możliwych miejsc: brzuch (lewa lub prawa strona) oraz przednia część uda (lewego lub prawego). Dawki > 30 µg na dobę (kolejne wstrzyknięcia) Wszystkie dawki > 30 µg na dobę należy podawać jako dwie pojedyncze dawki wstrzykiwane kolejno w różne miejsca wstrzyknięć (tabela 1).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Zaleca się stosowanie innego wstrzykiwacza produktu leczniczego Yorvipath do drugiego wstrzyknięcia danego dnia, nawet jeżeli dwa wstrzykiwacze mają przycisk tego samego koloru (taka sama moc). Tabela 1: Zalecany schemat dawkowania produktu leczniczego Yorvipath > 30 µg/dobę
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
Dawka Schemat dawkowania Połączenie wstrzykiwaczy 33 µg/dobę 15 µg/dobę + 18 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)* 36 µg/dobę 18 µg/dobę + 18 µg/dobę 39 µg/dobę 18 µg/dobę + 21 µg/dobę 42 µg/dobę 21 µg/dobę + 21 µg/dobę 45 µg/dobę 21 µg/dobę + 24 µg/dobę Jeden fabrycznie napełniony wstrzykiwacz produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)+Jeden fabrycznie napełniony produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk)** 48 µg/dobę 24 µg/dobę + 24 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk) 51 µg/dobę 24 µg/dobę + 27 µg/dobę 54 µg/dobę 27 µg/dobę + 27 µg/dobę 57 µg/dobę 27 µg/dobę + 30 µg/dobę 60 µg/dobę 30 µg/dobę + 30 µg/dobę - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDawkowanie
* Produkt leczniczy Yorvipath 294 mikrogramy/0,98 ml dostarcza dawki 15, 18 lub 21 µg PTH(1-34) (z pomarańczowym przyciskiem) ** Produkt leczniczy Yorvipath 420 mikrogramów/1,4 ml dostarcza dawki 24, 27 lub 30 µg PTH(1-34) (z bordowym przyciskiem)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1; - Pacjenci z rzekomą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkich zdarzeń hiperkalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe podczas rozpoczynania leczenia lub zwiększania dawki. W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy (patrz punkt 4.2) i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hiperkalcemii. Jeżeli wystąpi ciężka hiperkalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath (patrz punkt 4.2). Hipokalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkiej hipokalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe po nagłym odstawieniu leczenia, ale może wystąpić w dowolnym momencie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlSpecjalne środki ostrozności
W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii. Jeżeli wystąpi ciężka hipokalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath i dostosować stałe lub doraźne dawki czynnej witaminy D i (lub) suplementów wapnia (patrz punkt 4.2). Stosowanie jednocześnie z glikozydami nasercowymi Hiperkalcemia z dowolnego powodu może predysponować do toksyczności naparstnicy. U pacjentów stosujących produkt leczniczy Yorvipath jednocześnie z glikozydami nasercowymi (takimi jak digoksyna lub digitoksyna) należy monitorować stężenia wapnia oraz glikozydu nasercowego w surowicy, a pacjentów obserwować pod kątem objawów podmiotowych i przedmiotowych toksyczności naparstnicy (patrz punkt 4.5).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlSpecjalne środki ostrozności
Ciężka choroba nerek lub wątroby Nie przeprowadzono badań z udziałem pacjentów z ciężkimi zaburzeniami czynności nerek i ciężkimi zaburzeniami czynności wątroby. W przypadku tych pacjentów produkt leczniczy Yorvipath powinien być stosowany z zachowaniem ostrożności. Pacjenci z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min mogą być bardziej podatni na hiperkalcemię oraz przemijające obniżenie szacowanego współczynnika filtracji kłębuszkowej, szczególnie na początku leczenia. Jeżeli leczenie jest rozpoczynane u takich pacjentów, zaleca się monitorowanie stężeń wapnia w surowicy. Stosowanie u pacjentów ze zwiększonym ryzkiem kostniakomięsaka Produkt leczniczy Yorvipath nie był badany i powinien być stosowany z zachowaniem ostrożności u pacjentów: - z nowotworami szkieletu oraz przerzutami w kościach; - którzy otrzymują lub otrzymywali radioterapię układu kostnego; - z niewyjaśnionym wzrostem poziomu aktywności fosfatazy alkalicznej swoistej dla kości; - z chorobami metabolicznymi kości, którzy są narażeni na podwyższone bazowe ryzyko kostniakomięsaka (np.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlSpecjalne środki ostrozności
choroba kości Pageta). Stosowanie u pacjentów z osteoporozą Badania przesiewowe i monitorowanie pod kątem osteoporozy powinno być zgodne z lokalnymi praktykami klinicznymi w przypadku każdego pacjenta ze zwiększonym ryzykiem złamań z powodu kruchości kości (patrz punkt 4.8). Zawartość sodu Lek zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że produkt leczniczy uznaje się za „wolny od sodu”.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji Nie przeprowadzono badań dotyczących interakcji z innymi produktami leczniczymi. Glikozydy nasercowe (takie jak digoksyna lub digitoksyna) mają wąski wskaźnik terapeutyczny, a na ich działanie wpływa wapń. U pacjentów przyjmujących produkt leczniczy Yorvipath oraz glikozydy nasercowe należy obserwować, czy nie występują objawy przedmiotowe i podmiotowe toksyczności naparstnicy. Inne produkty lecznicze mogące wywierać wpływ na stężenie wapnia w surowicy oraz zmieniać odpowiedź terapeutyczną produktu leczniczego Yorvipath to między innymi bisfosfoniany, denosumab, romosozumab, tiazyd oraz diuretyki pętlowe, kortykosteroidy układowe oraz lit. U pacjentów leczonych jednocześnie tymi produktami leczniczymi należy obserwować zmiany stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Ciąża Nie ma danych lub istnieją tylko ograniczone dane dotyczące stosowania produktu leczniczego Yorvipath u kobiet w ciąży. Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na reprodukcję (patrz punkt 5.3). Jednak nie można wykluczyć zagrożenia dla kobiet w ciąży lub rozwijającego się płodu. Decyzja o rozpoczęciu lub odstawieniu leczenia produktem leczniczym Yorvipath w trakcie ciąży powinna uwzględniać stosunek możliwych zagrożeń do korzyści dla kobiet w ciąży. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet w ciąży z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Karmienie piersią Nie wiadomo, czy palopegteryparatyd przenika do mleka ludzkiego. Ponieważ palopegteryparatyd nie jest wchłaniany po podaniu doustnym, jest mało prawdopodobne, aby miał niekorzystny wpływ na dziecko karmione piersią.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWpływ na płodność, ciążę i laktację
Podejmując decyzję o zaprzestaniu karmienia piersią lub leczenia produktem leczniczym Yorvipath, należy wziąć pod uwagę korzyści z karmienia piersią dla dziecka i korzyści z leczenia dla kobiety. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet karmiących piersią z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Płodność Nie przeprowadzono badań wpływu palopegteryparatydu na płodność człowieka. Dane z badań na zwierzętach nie wskazują, aby podawanie palopegteryparatydu upośledzało płodność (patrz punkt 5.3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Produkt leczniczy Yorvipath nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednak u niektórych pacjentów obserwowano zawroty głowy, stan przedomdleniowy, omdlenia i (lub) niedociśnienie ortostatyczne. Tacy pacjenci powinni powstrzymywać się od prowadzenia pojazdów i obsługiwania maszyn do czasu ustąpienia objawów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi w badaniach klinicznych z użyciem palopegteryparatydu były reakcje w miejscu wstrzyknięcia (21,6%), ból głowy (18,7%) i parestezja (13,7%). Najpoważniejszą reakcją niepożądaną zgłaszaną w badaniach klinicznych była hiperkalcemia (1,40%). Tabelaryczna lista reakcji niepożądanych Tabela 2 przedstawia reakcje niepożądane u pacjentów leczonych palopegteryparatydem zidentyfikowane we wszystkich badaniach fazy II oraz fazy III przeprowadzonych z grupami kontrolnymi otrzymującymi placebo. Reakcje niepożądane wymienione w poniższej tabeli przedstawiono według klasyfikacji układów i narządów MedDRA oraz częstości występowania zdefiniowanej za pomocą następującej konwencji: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000) i nieznana (częstość występowania nie może być określona na podstawie dostępnych danych).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
W obrębie każdej kategorii częstości występowania reakcje niepożądane przedstawiono według malejącej ciężkości. Tabela 2: Częstość występowania reakcji niepożądanych palopegteryparatydu
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
Klasyfikacja układówi narządów MedDRA Częstość występowania Reakcja niepożądana Zaburzenia metabolizmui odżywiania często hiperkalcemiaa, hipokalcemia Zaburzenia układu nerwowego bardzo często ból głowyd, parestezjaa często zawroty głowya, c, d, omdlenied,stan przedomdleniowyd Zaburzenia serca często palpitacjed, zespół posturalnej tachykardii ortostatycznejd Zaburzenia naczyniowe często niedociśnienie ortostatyczned niezbyt często nadciśnieniee Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia często ból jamy ustnej i gardła Zaburzenia żołądka i jelit bardzo często nudnościa często biegunkaa, zaparcia, wymioty,dyskomfort w jamie brzusznej, ból w jamie brzusznej Zaburzenia skóry i tkanki podskórnej często wysypka, reakcja nadwrażliwości na światło Zaburzenia mięśniowo- szkieletowe i tkanki łącznej często ból stawów, mialgia, drżenie mięśnif, ból mięśniowo-szkieletowyf Zaburzenia nerek i dróg moczowych niezbyt często nokturiae częstość nieznana wielomocze Zaburzenia ogólne i stany w miejscu podania bardzo często reakcje w miejscu wstrzyknięciaa,b, zmęczenie często astenia, pragnienie niezbyt często dyskomfort w klatce piersiowejf, ból w klatce piersiowejf Badania diagnostyczne częstość nieznana gęstość kości zmniejszona - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
a W przypadku tych reakcji niepożądanych pierwsze wystąpienie pojawiało się niemal wyłącznie w ciągu pierwszych 3 miesięcy leczenia (okres dostosowywania dawki). b Reakcje w miejscu wstrzyknięcia obejmują reakcję w miejscu wstrzyknięcia, rumień w miejscu wstrzyknięcia, zasiniaczenie w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia, krwotok w miejscu wstrzyknięcia, wysypkę w miejscu wstrzyknięcia oraz opuchliznę w miejscu wstrzyknięcia. c Zawroty głowy obejmują zawroty głowy i posturalne zawroty głowy. d Objawy związane z rozszerzeniem naczyń krwionośnych obejmowały posturalne zawroty głowy, ból głowy, palpitacje, zespół posturalnej tachykardii ortostatycznej, niedociśnienie ortostatyczne, obniżone ortostatyczne ciśnienie krwi i omdlenie. Objawy związane z rozszerzeniem naczyń krwionośnych (zidentyfikowane w badaniach klinicznych) występowały częściej podczas pierwszych 3 miesięcy leczenia i stanowiły podzbiór wszystkich zdarzeń zgłaszanych jako reakcje niepożądane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
W badaniu TCP-304 w ciągu pierwszych 3 miesięcy wystąpiły łącznie 3 zdarzenia (u 2 pacjentów) uznane za związane z palopegteryparatydem: posturalne zawroty głowy (n=1) oraz ból głowy i palpitacje (n=1). e Objawy przedmiotowe i podmiotowe potencjalnie związane z hiperkalcemią, jak zaobserwowano w badaniach klinicznych. f Objawy przedmiotowe i podmiotowe potencjalnie związane z hipokalcemią, jak zaobserwowano w badaniach klinicznych. Opis wybranych reakcji niepożądanych Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki hiperkalcemii. Częstość występowania hiperkalcemii była wyższa u pacjentów leczonych produktem leczniczym Yorvipath w porównaniu z placebo. W trakcie okresu zaślepionego objawową hiperkalcemię zgłoszono u 8,6% pacjentów leczonych produktem leczniczym Yorvipath i wszystkie zgłoszone przypadki wystąpiły w ciągu pierwszych 3 miesięcy po rozpoczęciu leczenia produktem leczniczym Yorvipath.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
Immunogenność U pacjentów mogą pojawić się przeciwciała przeciwko palopegteryparatydowi. Odsetek pacjentów z dodatnim wynikiem testu na przeciwciała wiążące był niski w dowolnym momencie leczenia — 0,7% miało niskie miano przeciwciał nieneutralizujących przeciwko PTH, a 5% miało niskie miano związanych z leczeniem przeciwciał przeciwko PEG. U 2,2% pacjentów leczonych palopegteryparatydem z wcześniej nabytymi przeciwciałami przeciwko PEG zaobserwowano przejściowy wpływ na PK (zwiększony klirens całkowitego PTH, mPEG i zmniejszenie stężenia PTH) oraz zmniejszenie stężenia wapnia w surowicy. Jednakże skuteczność terapeutyczna była utrzymana dzięki odpowiedniemu dostosowaniu dawki palopegteryparatydu zgodnie z algorytmem dostosowywania dawki w badaniu. Reakcje w miejscu wstrzyknięcia Reakcje w miejscu wstrzyknięcia były najczęściej zgłaszanymi w badaniach klinicznych reakcjami niepożądanymi (mediana czasu wystąpienia wynosiła 2,5 dnia; częstość występowania 21,6%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
Najczęstszymi reakcjami w miejscu wstrzyknięcia były rumień miejscowy (wszystkie o średnicy < 5 cm, a większość od 0 do < 2 cm) o nasileniu łagodnym lub umiarkowanym (stopień 1 lub 2) z medianą czasu trwania 72 godziny. Wszystkie reakcje w miejscu wstrzyknięcia ustępowały bez leczenia, żadna nie była ciężka ani nie prowadziła do przerwania leczenia. Objawy związane z rozszerzeniem naczyń krwionośnych Podczas stosowania leku Yorvipath zgłaszano objawy rozszerzenia naczyń krwionośnych. Objawy te są zwykle przemijające i ustępują bez leczenia; żaden nie był poważny ani nie doprowadził do przerwania leczenia. W przypadku wystąpienia objawów zaleca się dawkowanie przed snem, w pozycji leżącej. Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDziałania niepożądane
Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzedawkowanie
4.9 Przedawkowanie W razie przedawkowania pacjent powinien być obserwowany przez fachowy personel medyczny. Przedawkowanie może powodować hiperkalcemię, której objawy mogą obejmować odwodnienie, palpitacje serca, zmiany EKG, niedociśnienie, nudności, wymioty, zawroty głowy, osłabienie mięśni oraz splątanie. Pacjent z ciężką hiperkalcemią może wymagać obserwacji i opieki medycznej (patrz punkt 4.4). Jeden przypadek około 3-krotnego przypadkowego przedawkowania przepisanej dawki trwającego ponad 7 kolejnych dni spowodował stężenie wapnia w surowicy wynoszące aż 16,1 mg/dl, pacjent miał objawy i wymagał interwencji medycznej. Po wstrzymaniu podawania palopegteryparatydu, wapnia oraz czynnej witaminy D pacjent wyzdrowiał i ponownie rozpoczął przyjmowanie prawidłowej dawki.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: Hemostaza wapnia, parathormony i analogi, kod ATC: H05AA05 Mechanizm działania Endogenny parathormon (PTH) jest wydzielany przez przytarczyce jako polipeptyd o długości 84 aminokwasów. PTH wywiera działanie za pośrednictwem receptorów parathormonu na powierzchni komórek, na przykład ulegającego ekspresji w tkance kości, nerek i nerwów. Aktywacja PTH1R stymuluje obrót kostny, zwiększa wchłanianie zwrotne wapnia i wydalanie fosforanów w nerkach oraz ułatwia syntezę czynnej witaminy D. Palopegteryparatyd jest prolekiem zawierającym PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez zastrzeżony łącznik TransCon. PTH(1-34) i jego główny metabolit, PTH(1-33) mają podobne powinowactwo oraz aktywują PTH1R jak endogenny PTH. W warunkach fizjologicznych PTH jest w kontrolowany sposób odszczepiany od palopegteryparatydu zapewniając ciągłą ekspozycję ogólnoustrojową na aktywny PTH.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Skuteczność kliniczna i bezpieczeństwo stosowania Badanie z udziałem pacjentów z ustaloną niedoczynnością przytarczyc W kluczowym badaniu klinicznym fazy III PaTHway (TCP-304) oceniano skuteczność i bezpieczeństwo stosowania produktu leczniczego Yorvipath jako terapii zastępującej PTH u osób dorosłych z niedoczynnością przytarczyc. 26-tygodniowy, prowadzony metodą podwójnie ślepej próby, kontrolowany placebo okres badania klinicznego obejmował pacjentów randomizowanych (3:1) do grupy przyjmującej produkt leczniczy Yorvipath w dawce początkowej 18 mikrogramów/dobę lub placebo podawane jednocześnie z terapią konwencjonalną (suplement wapnia i czynna witamina D). Randomizacja była stratyfikowana według etiologii niedoczynności przytarczyc (tj. po zabiegu chirurgicznym wobec wszystkich innych przyczyn).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Badane leczenie (palopegteryparatyd lub placebo) i terapia konwencjonalna były następnie dostosowywane według algorytmu dawkowania opartego na stężeniach wapnia w surowicy skorygowanych o stężenie albumin. Średnia wieku pacjentów podczas rekrutacji wynosiła 49 lat (od 19 do 78 lat; 12% miało ≥ 65 lat) i większość pacjentów była kobietami (78%) oraz rasy białej (93%). Osiemdziesiąt pięć procent (85%) miało niedoczynność przytarczyc nabytą po operacji szyi. Spośród pacjentów z innymi etiologiami niedoczynności przytarczyc 7 (8,5%) pacjentów miało chorobę idiopatyczną, 2 miało autoimmunologiczny zespół niedoczynności wielogruczołowej typu 1 (APS-1), 1 miał hipokalcemię autosomalną dominującą typu 1 (ADH1, mutacja CaSR), 1 miał zespół DiGeorge i 1 miał niedoczynności przytarczyc, głuchotę czuciowo-nerwową oraz zespół dysplazji nerek (HDR) (mutacja GATA3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Przed randomizacją wszyscy pacjenci zostali poddani trwającemu około 4 tygodnie okresowi przesiewowemu, podczas którego suplementy wapnia oraz czynnej witaminy D dostosowano, aby osiągnąć stężenie wapnia w surowicy skorygowane o stężenie albumin wynoszące od 1,95 do 2,64 mmol/l (od 7,8 do 10,6 mg/dl), stężenie magnezu ≥ 0,53 mmol/l (≥ 1,3 mg/dl) i poniżej górnego zakresu referencyjnego normy oraz stężenie 25(OH) witaminy D od 50 do 200 nmol/l (od 20 do 80 ng/ml). W przypadku terapii konwencjonalnej pacjenci byli leczeni średnim wyjściowymi dawkami wapnia (pierwiastkowego) wynoszącymi 1839 mg/dobę. Średnie wyjściowe dawki czynnej witaminy D wynosiły 0,75 mikrograma/dobę u pacjentów leczonych kalcytriolem (n=70) oraz 2,3 mikrograma/dobę u pacjentów leczonych alfakalcydolem (n=12). Średnie wyjściowe stężenie wapnia w surowicy skorygowane o stężenie albumin oraz średnie 24-godzinne stężenie wapnia w moczu były podobne w obu grupach leczenia: średnie stężenie wapnia w surowicy wynosiło 2,2 mmol/l (8,8 mg/dl) oraz 2,15 mmol/l (8,6 mg/dl), a średnie 24-godzinne stężenie wapnia w moczu wynosiło 392 mg/dobę i 329 mg/dobę odpowiednio w przypadku produktu leczniczego Yorvipath oraz placebo.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Pierwszorzędowy punkt końcowy Pierwszorzędowy złożony punkt końcowy był definiowany jako odsetek pacjentów w tygodniu 26, którzy osiągnęli: stężenia wapnia w surowicy w zakresie prawidłowym (od 2,07 do 2,64 mmol/l [od 8,3 do 10,6 mg/dl]), niezależnie od terapii konwencjonalnej definiowanej jako niewymagająca czynnej witaminy D oraz ≤ 600 mg/dobę suplementacji wapnia i bez zwiększenia przepisanego badanego leczenia w ciągu 4 tygodni przed tygodniem 26. Kluczowe drugorzędowe punkty końcowe obejmowały podzbiór punktacji domeny skali doświadczeń pacjentów z niedoczynnością przytarczyc (ang. Hypoparathyroidism Patient Experience Scale, HPES) oraz punktacji podskali skróconego formularza 36-elementowego (ang. 36-Item Short Form Survey, SF-36). Liczbę pacjentów spełniających pierwszorzędowy złożony punkt końcowy w porównaniu z grupą placebo i każdym składnikiem pierwszorzędowego punktu końcowego w tygodniu 26 przedstawiono w tabeli 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Tabela 3: TCP-304: współczynnik odpowiedzi oparty na pierwszorzędowym punkcie końcowym w tygodniu 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Yorvipath (N=61) (n, %) Placebo (N=21) (n, %) Różnica współczynnikaodpowiedzi (95% CI) Odpowiedź w tygodniu 26 48 (78,7%) 1 (4,8%) 74,0%(60,4%; 87,6%)p < 0,0001 Odpowiedź dla każdego składnika Stężenie wapnia w surowicy skorygowaneo stężenie albumin w zakresie prawidłowyma 49 (80,3%) 10 (47,6%) 32,7%(9,2%; 56,3%) Niezależność od czynnej witaminy Db 60 (98,4%) 5 (23,8%) 74,6%(56,1%; 93,1%) Niezależność od terapeutycznych dawek wapniac 57 (93,4%) 1 (4,8%) 88,7%(77,7%; 99,7%) Brak zwiększenia dawki w grupie produktu leczniczego Yorvipathd 57 (93,4%) 12 (57,1%) 36,4%(14,2%; 58,5%) - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
a Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosił od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl). b Wszystkie codzienne stałe dawki czynnej witaminy D równe zero ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. c Średnie codzienne stałe dawki pierwiastkowego wapnia ≤ 600 mg ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. d Brak zwiększenia dawki produktu leczniczego Yorvipath w ciągu 4 tygodni przed wizytą po 26 tygodniach. Skróty: CI: przedział ufności; PRN: pro re nata. Drugorzędowe punkty końcowe Przyjmowanie terapii konwencjonalnej: dawki wapnia i czynnej witaminy D. W badaniu fazy III PaTHway, w tygodniu 26, 93% (57/61) pacjentów w grupie produktu leczniczego Yorvipath było w stanie odstawić terapię konwencjonalną (tj. odstawić czynną witaminę D oraz terapeutyczne dawki wapnia).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Wszyscy pacjenci w grupie produktu leczniczego Yorvipath odstawili czynną witaminę D do tygodnia 8 i utrzymywali zmniejszenie terapeutycznych dawek wapnia. Występowało znaczące zmniejszenie przyjmowania terapii konwencjonalnej w grupie produktu leczniczego Yorvipath od wartości wyjściowej do tygodnia 26 w porównaniu z placebo: czynnej witaminy D (nominalna wartość p < 0,0001), dawki wapnia (nominalna wartość p = 0,0003) oraz codziennego obciążenia tabletkami (nominalna wartość p < 0,0001) (tabela 4). Tabela 4: Drugorzędowe punkty końcowe: przyjmowanie terapii konwencjonalnej w tygodniu 26 — okres zaślepiony (populacja ITT)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Yorvipath (n/N=60/61)a Placebo (n/N=19/21)a Nominalnawartość p Wartość początkowa Tydzień 26 Wartość początkowa Tydzień 26 Suplementacyjna dawkaczynnej witaminy D (µg), średnia (SD) 1,0 (0,7) 0,0 (0,0) 1,0 (0,6) 0,6 (0,7) < 0,0001 Suplementacyjna dawka wapnia (mg), średnia (SD) 1737 (907) 274 (177) 2089 (1448) 1847 (1326) 0,0003 Codzienne obciążenie tabletkami (liczba tabletekterapii konwencjonalnej), średnia (SD) 6,6 (2,1) 0,5 (1,7) 6,3 (2,8) 5,4 (3,2) < 0,0001 - CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Nominalna wartość p z badań różnic zmiany od wartości wyjściowej do tygodnia 26 między produktem leczniczym Yorvipath oraz placebo. a N to liczba pacjentów w populacji ITT; n to liczba pacjentów z danymi zarówno wyjściowymi, jak i w tygodniu 26. Parametry biochemiczne surowicy Średnia wartość wapnia w surowicy początkowo wzrosła i utrzymywała się w zakresie prawidłowym u pacjentów leczonych palopegteryparatydem (rysunek 2). U pacjentów przyjmujących placebo stężenia wapnia w surowicy nieco zmniejszyły się, spadając poniżej zakresu prawidłowego w tygodniu 2 (średnia obserwowana wartość: 2,06 mmol/l) i w tygodniu 26 (średnia obserwowana wartość: 2,06 mmol/l). Różnica średniej najmniejszych kwadratów leczenia między produktem leczniczym Yorvipath i placebo wynosiła 0,17 mmol/l (95% CI: 0,100; 0,247; nominalna wartość p < 0,0001) w tygodniu 26. Rysunek 2: Wapń w surowicy (średnia ±SE) według wizyty — okres zaślepiony (populacja ITT) 3 2,5 2 1,5 0 2 4 6 8 10 12 14 16 18 20 22 24 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Średnie (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mmol/l) Czas (tygodnie)
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Palopegteryparatyd
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Placebo Średnie stężenia fosforanu w surowicy u pacjentów leczonych palopegteryparatydem były w zakresie prawidłowym wyjściowo i utrzymywały się w zakresie prawidłowym do tygodnia 26 (średnia zmiana od wartości wyjściowej do tygodnia 26 wynosiła -0,13 mmol/l). Średni iloczyn wapnia i fosforanów w surowicy zmniejszył się u pacjentów leczonych produktem leczniczym Yorvipath i pozostawał stabilny w zakresie prawidłowym do tygodnia 26. 24-godzinne wydalanie wapnia w moczu Terapia produktem leczniczym Yorvipath normalizowała 24-godzinną średnią wydalania wapnia w moczu i wykazywała większe zmniejszenie 24-godzinnego stężenia wapnia w moczu niż placebo. Dzieci i młodzież Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego Yorvipath w jednej lub kilku podgrupach populacji dzieci i młodzieży z niedoczynnością przytarczyc, zgodnie z warunkami zawartymi w decyzji dotyczącej planu badań dzieci i młodzieży (PIP, ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakodynamiczne
Paediatric Investigation Plan) we wskazaniu leczenia niedoczynności przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Wchłanianie Po codziennym podawaniu podskórnym palopegteryparatyd uwalnia PTH za pośrednictwem autoodszczepienia łącznika TransCon podlegając kinetyce pierwszorzędowej, dając ciągłą ekspozycję przez 24 godziny w szacowanym zakresie prawidłowym (rysunek 3). Rysunek 3: Średni uwalniany PTH* po podawaniu podskórnym palopegteryparatydu w stanie stacjonalnym u pacjentów z niedoczynnością przytarczyc 40 30 20 10 0 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
Średni (+/-SE) uwalniany PTH (pg/ml) Czas (godz.) Szacowany zakres prawidłowy dla PTH(1-34) wynosi około 4 do 26 pg/ml. Jest on obliczony na podstawie PTH(1-34) stanowiącego 40% masy cząsteczkowej PTH(1-84)** i zakresu prawidłowego (10 do 65 pg/ml) dla PTH(1-84). * Średnia dawka palopegteryparatydu (zakres): 22,3 (12-33) µg/dobę, n=7. uwolniony PTH: suma PTH(1-34) i PTH(1-33). ** PTH(1-84) = endogenny parathormon. U pacjentów z niedoczynnością przytarczyc podawanie palopegteryparatydu odpowiadało 18 µg PTH(1-34)/dobę, przewidywane maksymalne stężenie w osoczu (C max ) (CV%) palopegteryparatydu wynosiło 5,18 ng/ml (36%), a przewidywana wartość C max (CV%) dla uwalnianego PTH wynosiła 6,9 pg/ml (22%) z medianą czasu do osiągnięcia maksymalnych stężeń (T max ) wynoszącą 4 godziny. Przewidywana ekspozycja w ciągu 24-godzinnego odstępu między dawkami (pole pod krzywą, ang. area under the curve, AUC) (CV%) dla uwalnianego PTH wynosiła 150 pg*h/ml (22%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
Po podaniu wielu podskórnych dawek palopegteryparatydu w zakresie od 12 do 24 µg PTH(1-34)/dobę stężenia palopegteryparatydu i uwalnianego PTH rosły w sposób proporcjonalny do dawki, osiągając stan stacjonarny w ciągu odpowiednio około 10 i 7 dni. Stosunek wartości maksymalnej do minimalnej był niski, wynosił około 1,1 oraz 1,5 przez 24 godziny stanu stacjonarnego odpowiednio dla palopegteryparatydu i uwalnianego PTH. Palopegteryparatyd ulegał akumulacji po wielu dawkach do 18-krotności wartości AUC. Dystrybucja Pozorną objętość dystrybucji (CV%) palopegteryparatydu szacuje się na 4,8 l (50%) oraz do 8,7 l (18%) dla uwalnianego PTH. Metabolizm PTH uwalniany z palopegteryparatydu składa się z PTH(1-34) i metabolitu PTH(1-33). PTH jest metabolizowany i usuwany w nerkach. Eliminacja U zdrowych osób dorosłych klirens (CV%) palopegteryparatydu w stanie stacjonarnym szacuje się na 0,58 l/dobę (52%) z przewidywanym okresem półtrwania wynoszącym 70 godzin.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
Pozorny okres półtrwania PTH uwalnianego z palopegteryparatydu wynosi około 60 godzin. W wątrobie większość PTH jest rozszczepiana przez katepsyny. W nerkach niewielka ilość PTH wiąże się z PTH1R, ale większość jest wydalana poprzez filtrację kłębuszkową. Zależności farmakokinetyczno-farmakodynamiczne W cząstkowym badaniu farmakodynamiki/farmakokinetyki u pacjentów z niedoczynnością przytarczyc, codzienne podskórne podawanie palopegteryparatydu (średnia dawka (zakres): 22,3 (12– 33) µg PTH (1-34)dobę) zwiększała stężenie wapnia w surowicy do zakresu prawidłowego (patrz rysunek 4). Zwiększenie stężeń wapnia w surowicy następowało w sposób zależny od dawki wspierając możliwość dostosowania dawki palopegteryparatydu odpowiednio do zmierzonych wartości wapnia w surowicy u danego pacjenta. mmol/l Rysunek 4: Średnie stężenia wapnia w surowicy skorygowane o stężenie albumin po podawaniu podskórnym palopegteryparatydu w stanie stacjonarnym u pacjentów z niedoczynnością przytarczyc 12 11 10 9 8 7 3 2,75 2,5 2,25 2 1,75 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
Średnia (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mg/dl) Czas (godz.) Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosi od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl), co oznaczono szarym cieniowaniem. Średnia dawka palopegteryparatydu (zakres): 22,3 (12–33) µg PTH(1- 34)/dobę, n=7. Szczególne grupy pacjentów Na farmakokinetykę uwalnianego PTH nie wpływa płeć ani masa ciała. Dane dotyczące rasy i pochodzenia etnicznego nie wykazują żadnych trendów wskazujących na różnice, ale dostępne dane są zbyt ograniczone do postawienia rozstrzygających wniosków. Osoby w starszym wieku Na farmakokinetykę uwalnianego PTH nie wpływa wiek (od 19 do 76 lat). Zaburzenia czynności nerek Produkt leczniczy Yorvipath podawano pacjentom z niedoczynnością przytarczyc z eGFR ≥ 30 ml/min w długotrwałych badaniach klinicznych bez konieczności dostosowywania dawki poza algorytmem dostosowywania dawki w ramach badania.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlWłaściwości farmakokinetyczne
Nie przeprowadzono badań klinicznych z udziałem pacjentów z niedoczynnością przytarczyc oraz zaburzeniem czynności nerek (< 30 ml/min) lub dializowanych. W badaniu, w którym produkt leczniczy Yorvipath podawano jako pojedynczą dawkę uczestnikom bez niedoczynności przytarczyc, ekspozycja na palopegteryparatyd i uzyskiwane stężenia wapnia w surowicy były podobne jak u uczestników z łagodnym, umiarkowanym lub ciężkim upośledzeniem czynności nerek w porównaniu do pacjentów bez zaburzeń czynności nerek.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie Nie ujawniono szczególnego zagrożenia dla człowieka wynikającego z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, genotoksyczności i tolerancji miejscowej przeprowadzonych na palopegteryparatydzie. U zwierząt wszystkich zastosowanych gatunków podanie wielokrotne największych dawek powodowało niepożądaną uporczywą hiperkalcemię, która w niektórych badaniach prowadziła do przedwczesnego zgonu/eutanazji, objawów klinicznych, utrata masy ciała i (lub) mineralizacji tkanki miękkiej obserwowanej głównie w nerkach. Wyniki te są uważane za skutki nieustannie przesadnej farmakologii PTH i nie mają one znaczenia w warunkach klinicznych, gdzie dawkę dostosowuje się w celu zapewnienia normalizacji stężenia wapnia w surowicy Zgodnie z oczekiwanymi skutkami farmakologicznymi, powtarzane codzienne podawanie palopegteryparatydu zwiększało obrót kostny u szczurów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzedkliniczne dane o bezpieczeństwie
Przy małych dawkach (2-krotność maksymalnej zalecanej dawki dla człowieka, MRHD) na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny indukował netto całkowite kataboliczne skutki kostne. Przy dużych dawkach (5-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny powodował netto anaboliczny skutek kostny. Dysplazja nasad kości była obserwowana przy najwyższej dawce (9-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów. Skutki te nie mają znaczenia w warunkach klinicznych, w których dawki leku Yorvipath dostosowuje się indywidualnie. Nie było skutków sercowo-naczyniowych u małp do najwyższej badanej dawki (włącznie) w badaniach dawki pojedynczej (3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ) lub dawki powtarzanej (0,98-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ).
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzedkliniczne dane o bezpieczeństwie
Nasilone występowanie kostniakomięsaków obserwowano w badaniach rakotwórczości z krótko żyjącymi analogami PTH u szczurów, ale bez dowodów zwiększonego ryzyka kostniakomięsaka u pacjentów leczonych krótko żyjącymi analogami PTH.Dlatego też nie przeprowadzono żadnego badania nad rakotwórczością palopegteryparatydu. W badaniach reprodukcji na zwierzętach podawanie palopegteryparatydu ciężarnym szczurom i królikom w okresie organogenezy nie dawało dowodów na letalność zarodka, toksyczność dla płodu lub dysmorfogenezę w przypadku podania najwyższych badanych dawek (włącznie) (odpowiednio 8- i 7-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC). Skutki przesadnej farmakologii PTH obserwowano przy najwyższych badanych dawkach u ciężarnych szczurów oraz królików (podwyższone stężenia wapnia w surowicy, zmniejszona masa ciała, zmniejszone spożycie pokarmu i (lub) oznaki kliniczne). Ekspozycje przy poziomie dawkowania, przy którym nie obserwuje się szkodliwych zmian (ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlPrzedkliniczne dane o bezpieczeństwie
no observed adverse effect level, NOAEL) w przypadku toksyczności dla matki wynosił 2- i 3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC u odpowiednio ciężarnych szczurów i królików. Nie przeprowadzono badania rozwoju pre- i postnatalnego związanego z palopegteryparatydem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Kwas bursztynowy Mannitol Metakrezol Sodu wodorotlenek Kwas chlorowodorowy (do regulacji pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 3 lata. Po pierwszym otwarciu Przechowywać w temperaturze poniżej 30 °C. Pozostawić zatyczkę wstrzykiwacza na fabrycznie napełnionym wstrzykiwaczu w celu ochrony przed światłem. Produkt leczniczy Yorvipath należy wyrzucić po 14 dniach. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2 °C – 8 °C). Nie zamrażać. Przechowywać w oryginalnym opakowaniu z założoną zatyczką wstrzykiwacza w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po pierwszym otwarciu, patrz punkt 6.3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDane farmaceutyczne
6.5 Rodzaj i zawartość opakowania Wkład (szkło typu 1) z tłokiem (halobutyl) i laminowanym arkuszem gumowym (halobutyl/izopren) umieszczony w wielodawkowym jednorazowym fabrycznie napełnionym wstrzykiwaczu wykonanym z polipropylenu. Opakowania dwóch fabrycznie napełnionych wstrzykiwaczy oraz 30 igieł jednorazowych na 28 dni leczenia (zapakowane razem w wewnętrznych pudełkach). Każde wewnętrzne pudełko zawiera jeden fabrycznie napełniony wstrzykiwacz oraz 15 igieł na 14 dni leczenia. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 6, 9 lub 12 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest niebieski.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDane farmaceutyczne
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 15, 18 lub 21 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest pomarańczowy. Yorvipath 420 mikrogramy/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 24, 27 lub 30 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest bordowy. 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Przygotowanie dawki Nowy wstrzykiwacz produktu leczniczego Yorvipath należy wyjąć z lodówki na 20 minut przed pierwszym otwarciem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDane farmaceutyczne
Roztwór powinien być przezroczysty, bezbarwny i wolny od cząstek stałych. Nie należy wstrzykiwać produktu leczniczego, jeżeli jest mętny lub zawiera cząstki stałe. Każdy fabrycznie napełniony wstrzykiwacz jest przeznaczony do użytku przez jednego pacjenta. Fabrycznie napełniony wstrzykiwacz nie powinien być współdzielony przez pacjentów, nawet jeżeli igła jest zmieniana. Jeżeli fabrycznie napełniony wstrzykiwacz był zamrażany lub narażony na ciepło, należy go wyrzucić. Za każdym razem, gdy fabrycznie napełniony wstrzykiwacz jest przygotowywany do podawania, należy założyć nową igłę. Igieł nie wolno używać ponownie. Może to zapobiec zablokowaniu igieł, zanieczyszczeniu, zakażeniu, wyciekowi roztworu lub niedokładnemu dawkowaniu. Igła iniekcyjna powinna być zdejmowana po każdym wstrzyknięciu, a wstrzykiwacz należy przechowywać bez przymocowanej igły. Igłę należy wyrzucać po każdym wstrzyknięciu.
- CHPL leku Yorvipath, roztwór do wstrzykiwań we wstrzykiwaczu, 294 mcg/0,98 mlDane farmaceutyczne
Instrukcje przygotowywania i podawania produktu leczniczego Yorvipath podano w ulotce dołączonej do opakowania oraz instrukcji użycia. Utylizacja Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Produkt leczniczy Yorvipath zawiera PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez łącznik. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 6, 9 lub 12 mikrogramów PTH(1-34).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 15, 18 lub 21 mikrogramów PTH(1-34). Yorvipath 420 mikrogramów/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika*. Stężenie oparte na zawartości PTH(1-34) wynosi 0,3 mg/ml. Każdy fabrycznie napełniony wstrzykiwacz półautomatyczny dostarcza dawki 24, 27 lub 30 mikrogramów PTH(1-34). * Moc wskazuje ilość PTH(1-34) bez uwzględniania łącznika mPEG. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlNazwa produktu leczniczego, skład i postać farmaceutyczna
POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (płyn do wstrzykiwań) Przezroczysty i bezbarwny o pH 3,7–4,3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWskazania do stosowania
4.1 Wskazania do stosowania Produkt leczniczy Yorvipath to terapia zastępcza parathormonem (PTH) wskazana do leczenia dorosłych z przewlekłą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
4.2 Dawkowanie i sposób podawania Leczenie powinno być rozpoczęte i monitorowane przez lekarzy lub wykwalifikowany personel medyczny doświadczony w diagnostyce oraz leczeniu pacjentów z niedoczynnością przytarczyc. Dawkowanie Zalecene dawki produktu leczniczego Yorvipath odnoszą się do mikrogramów PTH(1-34). Dawka powinna być dostosowana indywidualnie na podstawie stężenia wapnia w surowicy. Optymalna dawka po dostosowaniu to minimalna dawka wymagana do zapobiegania hipokalcemii. Jest to dawka, która utrzymuje stężenie wapnia w surowicy w prawidłowym zakresie bez potrzeby suplementacji czynnymi formami witaminy D lub wapniem w zakresie prawidłowym wykraczającym poza zalecaną suplementację żywieniową dla populacji ogólnej (ogólnie mniej niż 600 mg na dobę). Dawki czynnych postaci witaminy D i suplementów wapnia będą wymagać dostosowania przed rozpoczęciem i w trakcie leczenia produktem leczniczym Yorvipath na podstawie wartości wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Pacjenci otrzymujący maksymalną dawkę produktu leczniczego Yorvipath wynoszącą 60 µg na dobę, którzy doświadczają ciągłej hipokalcemii, mogą wymagać jednoczesnego podawania terapeutycznego wapnia i (lub) czynnych postaci witaminy D. Przed rozpoczęciem podawania produktu leczniczego Yorvipath Stężenie 25(OH) witaminy D w surowicy powinno być w zakresie prawidłowym, a stężenie wapnia w surowicy powinno być stabilne w zakresie prawidłowym lub nieco poniżej zakresu prawidłowego (1,95–2,64 mmol/l [7,8–10,6 mg/dl]) na co najmniej 1 wartość laboratoryjną na co najmniej dwa tygodnie przed pierwszą dawką leczenia. Rozpoczęcie podawania produktu leczniczego Yorvipath Zalecana dawka początkowa wynosi 18 µg raz na dobę ze stopniowym dostosowaniem dawki o 3 µg co 7 dni (patrz rysunek 1). Zakres dawki wynosi od 6 do 60 µg na dobę. Podczas rozpoczynania leczenia produktem leczniczym Yorvipath dawki czynnych postaci witaminy D lub suplementów wapnia powinny zostać dostosowane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
W razie przyjmowania czynnej witaminy D: o Jeżeli stężenie wapnia w surowicy wynosi ≥ 2,07 mmol/l [≥ 8,3 mg/dl/l], czynną witaminę D (kalcytriol lub alfakalcydol) należy odstawić tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. o Jeżeli stężenie wapnia w surowicy wynosi < 2,07 mmol/l [< 8,3 mg/dl/l], czynną witaminę D należy zmniejszyć o ≥ 50% tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. Dawki suplementów wapnia należy utrzymywać. W razie nieprzyjmowania czynnej witaminy D: o Suplementy wapnia należy zmniejszyć o co najmniej 1500 mg tego samego dnia, gdy podana jest pierwsza dawka produktu leczniczego Yorvipath. W razie przyjmowania dawek wapnia ≤ 1500 mg na dobę suplementy wapnia należy odstawić całkowicie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Jeżeli suplementy wapnia są wskazane w celu spełnienia wymogów dietetycznych, można rozważyć dalsze przyjmowanie suplementów wapnia w dawkach ≤ 600 mg na dobę zamiast ich całkowitego odstawienia. Dostosowanie i podtrzymywanie dawki produkt leczniczego Yorvipath Stężenie wapnia w surowicy musi być monitorowane w trakcie dostosowywania dawki (patrz punkt 4.4). Dawkę produktu leczniczego Yorvipath można stopniowo zwiększać o 3 µg, jeżeli upłynęło co najmniej 7 dni od wcześniejszej zmiany dawki (patrz rysunek 1). Nie wolno zwiększać dawki częściej niż co 7 dni. Dawkę produktu leczniczego Yorvipath można stopniowo zmniejszać o 3 µg nie częściej niż co 3 dni w odpowiedzi na hiperkalcemię (patrz rysunek 1). Stężenie wapnia w surowicy należy oznaczyć 7 dni po pierwszej dawce, oraz należy przestrzegać prawidłowego dawkowania produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementu wapnia zgodnie z zasadami przedstawionymi na rysunku 1.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Po każdej kolejnej zmianie dawki produktu leczniczego Yorvipath, czynnej witaminy D lub suplementów wapnia należy oznaczyć stężenie wapnia w surowicy w ciągu od 7 do 14 dni; pacjenci powinni być monitorowani pod kątem objawów klinicznych hipokalcemii lub hiperkalcemii. Dawki produktu leczniczego Yorvipath, czynnej witaminy D i (lub) suplementów wapnia należy dostosować zgodnie z rysunkiem 1. Dostosowywanie dawek produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia należy dokonywać tego samego dnia. Dawka podtrzymująca powinna być dawką, która zapewnia stężenie wapnia w surowicy w zakresie prawidłowym bez potrzeby suplementacji czynną witaminą D lub dawkami terapeutycznymi wapnia. Opcjonalnie można kontynuować suplementację wapniem wystarczającą do spełnienia wymogów dietetycznych (≤ 600 mg na dobę). Stężenie wapnia i 25(OH) witaminy D w surowicy należy oznaczać zgodnie ze standardem opieki po osiągnięciu dawki podtrzymującej.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Suplementacja 25(OH) witaminą D (nieczynną witaminą D) może być konieczna w celu osiągnięcia prawidłowych stężeń w surowicy. Rysunek 1: Dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia Małe stężenie wapnia w surowicy (< 2,07 mmol/l [< 8,3 mg/dl]): Tak ≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Kontynuować te same dawki suplementu wapnia i czynnej witaminy D na podstawie oceny klinicznej lekarza prowadzącego i zwiększyć tę samą dawkę produktu leczniczego Yorvipath o 3 µg Zwiększyć dawki suplementów wapnia i (lub) czynnej witaminy D do wcześniejszych dawek na podstawie oceny klinicznej lekarza prowadzącego i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Stężenie wapnia w surowicy w normie (od ≥ 2,07 do < 2,64 mmol/l [od ≥ 8,3 do < 10,6 mg/dl]): Nie Tak Dalsze przyjmowanie czynnej witaminy D?
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Kontynuować te same dawki produktu leczniczego Yorvipath, suplementu wapnia i czynnej witaminy D Zmniejszyć lub odstawić czynną witaminę D i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Tak Dalsze przyjmowanie suplementów wapnia? Kontynuować tę samą dawkę produktu leczniczego Yorvipath Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć suplementy wapnia o ≥ 1500 mg i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg Odstawić suplementy wapnia i zwiększyć dawkę produktu leczniczego Yorvipath o 3 µg
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
≥ 7 dni od rozpoczęcia podawania lub zmiany dawki produktu leczniczego Yorvipath? Tak - CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Nie Nie Nie Duże stężenie wapnia w surowicy (od ≥ 2,65 do < 3,00 mmol/l [od ≥ 10,7 do < 12,0 mg/dl]): Tak Dalsze przyjmowanie czynnej witaminy D? Dalsze przyjmowanie suplementów wapnia? Zmniejszyć lub odstawić czynną witaminę D i kontynuować te same dawki produktu leczniczego Yorvipath oraz suplementów wapnia Nie Nie Tak Tak Czy suplement wapnia wynosi ≥ 1 500 mg/ dobę? Zmniejszyć dawkę produktu leczniczego Yorvipath o 3 µg Zmniejszyć suplementy wapnia o ≥ 1500 mg i kontynuować tę samą dawkę produktu leczniczego Yorvipath Odstawić suplementy wapnia i kontynuować tę samą dawkę produktu leczniczego Yorvipath Nie Bardzo duże stężenie wapnia w surowicy (≥ 3,00 mmol/l [≥ 12 mg/dl]): Leczenie należy wstrzymać na 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Jeżeli kolejne stężenie wapnia w surowicy jest < 3,00 mmol/l [< 12 mg/dl], należy wznowić dostosowywanie dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z odpowiednim punktem rysunku 1, stosując najnowszą uzyskaną wartość stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Jeżeli stężenie wapnia w surowicy pozostaje ≥ 3,00 mmol/l [≥ 12 mg/dl], należy wstrzymać podawanie produktu leczniczego Yorvipath na dodatkowe 2 do 3 dni, a następnie ponownie sprawdzić stężenie wapnia w surowicy. Więcej informacji na temat hiperkalcemii, patrz punkt 4.4. Pominięta dawka Jeśli pominięto dawkę o mniej niż 12 godzin, należy ją podać jak najszybciej. Jeśli dawkę pominięto o więcej niż 12 godzin, należy ją pominąć, a następną dawkę podać zgodnie z planem. Przerwanie lub odstawienie produktu leczniczego Yorvipath Należy unikać przerywania codziennego podawania w celu zminimalizowania wahań stężenia PTH w surowicy. Przerwanie lub odstawienie leczenia może spowodować hipokalcemię. Po przerywaniu lub odstawieniu leczenia na 3 lub więcej kolejnych dni należy obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii oraz rozważyć oznaczenie wapnia w surowicy. Jeżeli jest to wskazane, należy wznowić leczenie suplementami wapnia i czynną witaminą D.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Wznowić leczenie przepisaną dawką jak najszybciej po przerwaniu. Podczas wznawiania leczenia po przerwie oznaczyć stężenie wapnia w surowicy i dostosować dawki produktu leczniczego Yorvipath, czynnej witaminy D oraz suplementów wapnia zgodnie z rysunkiem 1. Szczególne grupy pacjentów Osoby w starszym wieku Nie jest wymagane dostosowanie dawki zależnie od wieku (patrz punkt 5.2). Zaburzenie czynności wątroby Produkt leczniczy Yorvipath nie był badany u pacjentów z ciężkimi zaburzeniami czynności wątroby i u tych pacjentów powinien być stosowany z zachowaniem ostrożności (patrz punkt 4.4). Zaburzenie czynności nerek Nie jest wymagane dostosowanie dawki u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej (eGFR) ≥ 30 ml/min. W przypadku stosowania produktu leczniczego Yorvipath u pacjentów z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min zalecane jest częstsze oznaczanie stężenia wapnia w surowicy (patrz punkt 4.4).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Produkt leczniczy Yorvipath nie był badany u pacjentów z niedoczynnością przytarczyc oraz ciężkimi zaburzemiami czynności nerek (szacowany współczynnik filtracji kłębuszkowej < 30 ml/min) (patrz punkt 5.2). Dzieci i młodzież Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności produktu leczniczego Yorvipath u dzieci w wieku poniżej 18 lat. Dane nie są dostępne. Sposób podawania Produkt leczniczy Yorvipath musi być wstrzyknięty podskórnie w brzuch lub przednią część uda. Miejsca wstrzyknięcia należy codziennie zmieniać, wybierając jedno z czterech możliwych miejsc: brzuch (lewa lub prawa strona) oraz przednia część uda (lewego lub prawego). Dawki > 30 µg na dobę (kolejne wstrzyknięcia) Wszystkie dawki > 30 µg na dobę należy podawać jako dwie pojedyncze dawki wstrzykiwane kolejno w różne miejsca wstrzyknięć (tabela 1).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Zaleca się stosowanie innego wstrzykiwacza produktu leczniczego Yorvipath do drugiego wstrzyknięcia danego dnia, nawet jeżeli dwa wstrzykiwacze mają przycisk tego samego koloru (taka sama moc). Tabela 1: Zalecany schemat dawkowania produktu leczniczego Yorvipath > 30 µg/dobę
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
Dawka Schemat dawkowania Połączenie wstrzykiwaczy 33 µg/dobę 15 µg/dobę + 18 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)* 36 µg/dobę 18 µg/dobę + 18 µg/dobę 39 µg/dobę 18 µg/dobę + 21 µg/dobę 42 µg/dobę 21 µg/dobę + 21 µg/dobę 45 µg/dobę 21 µg/dobę + 24 µg/dobę Jeden fabrycznie napełniony wstrzykiwacz produktu leczniczego Yorvipath 294 µg/0,98 ml (pomarańczowy przycisk)+Jeden fabrycznie napełniony produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk)** 48 µg/dobę 24 µg/dobę + 24 µg/dobę Dwa fabrycznie napełnione wstrzykiwacze produktu leczniczego Yorvipath 420 µg/1,4 ml(bordowy przycisk) 51 µg/dobę 24 µg/dobę + 27 µg/dobę 54 µg/dobę 27 µg/dobę + 27 µg/dobę 57 µg/dobę 27 µg/dobę + 30 µg/dobę 60 µg/dobę 30 µg/dobę + 30 µg/dobę - CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDawkowanie
* Produkt leczniczy Yorvipath 294 mikrogramy/0,98 ml dostarcza dawki 15, 18 lub 21 µg PTH(1-34) (z pomarańczowym przyciskiem) ** Produkt leczniczy Yorvipath 420 mikrogramów/1,4 ml dostarcza dawki 24, 27 lub 30 µg PTH(1-34) (z bordowym przyciskiem)
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1; - Pacjenci z rzekomą niedoczynnością przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkich zdarzeń hiperkalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe podczas rozpoczynania leczenia lub zwiększania dawki. W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy (patrz punkt 4.2) i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hiperkalcemii. Jeżeli wystąpi ciężka hiperkalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath (patrz punkt 4.2). Hipokalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki ciężkiej hipokalcemii (patrz punkt 4.8). Ryzyko hiperkalcemii jest największe po nagłym odstawieniu leczenia, ale może wystąpić w dowolnym momencie.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlSpecjalne środki ostrozności
W trakcie leczenia należy oznaczyć stężenie wapnia w surowicy i obserwować, czy u pacjentów nie występują objawy przedmiotowe i podmiotowe hipokalcemii. Jeżeli wystąpi ciężka hipokalcemia, leczenie powinno być zgodne z wytycznymi klinicznymi i należy rozważyć dostosowanie dawki produktu leczniczego Yorvipath i dostosować stałe lub doraźne dawki czynnej witaminy D i (lub) suplementów wapnia (patrz punkt 4.2). Stosowanie jednocześnie z glikozydami nasercowymi Hiperkalcemia z dowolnego powodu może predysponować do toksyczności naparstnicy. U pacjentów stosujących produkt leczniczy Yorvipath jednocześnie z glikozydami nasercowymi (takimi jak digoksyna lub digitoksyna) należy monitorować stężenia wapnia oraz glikozydu nasercowego w surowicy, a pacjentów obserwować pod kątem objawów podmiotowych i przedmiotowych toksyczności naparstnicy (patrz punkt 4.5).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlSpecjalne środki ostrozności
Ciężka choroba nerek lub wątroby Nie przeprowadzono badań z udziałem pacjentów z ciężkimi zaburzeniami czynności nerek i ciężkimi zaburzeniami czynności wątroby. W przypadku tych pacjentów produkt leczniczy Yorvipath powinien być stosowany z zachowaniem ostrożności. Pacjenci z szacowanym współczynnikiem filtracji kłębuszkowej < 45 ml/min mogą być bardziej podatni na hiperkalcemię oraz przemijające obniżenie szacowanego współczynnika filtracji kłębuszkowej, szczególnie na początku leczenia. Jeżeli leczenie jest rozpoczynane u takich pacjentów, zaleca się monitorowanie stężeń wapnia w surowicy. Stosowanie u pacjentów ze zwiększonym ryzkiem kostniakomięsaka Produkt leczniczy Yorvipath nie był badany i powinien być stosowany z zachowaniem ostrożności u pacjentów: - z nowotworami szkieletu oraz przerzutami w kościach; - którzy otrzymują lub otrzymywali radioterapię układu kostnego; - z niewyjaśnionym wzrostem poziomu aktywności fosfatazy alkalicznej swoistej dla kości; - z chorobami metabolicznymi kości, którzy są narażeni na podwyższone bazowe ryzyko kostniakomięsaka (np.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlSpecjalne środki ostrozności
choroba kości Pageta). Stosowanie u pacjentów z osteoporozą Badania przesiewowe i monitorowanie pod kątem osteoporozy powinno być zgodne z lokalnymi praktykami klinicznymi w przypadku każdego pacjenta ze zwiększonym ryzykiem złamań z powodu kruchości kości (patrz punkt 4.8). Zawartość sodu Lek zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że produkt leczniczy uznaje się za „wolny od sodu”.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji Nie przeprowadzono badań dotyczących interakcji z innymi produktami leczniczymi. Glikozydy nasercowe (takie jak digoksyna lub digitoksyna) mają wąski wskaźnik terapeutyczny, a na ich działanie wpływa wapń. U pacjentów przyjmujących produkt leczniczy Yorvipath oraz glikozydy nasercowe należy obserwować, czy nie występują objawy przedmiotowe i podmiotowe toksyczności naparstnicy. Inne produkty lecznicze mogące wywierać wpływ na stężenie wapnia w surowicy oraz zmieniać odpowiedź terapeutyczną produktu leczniczego Yorvipath to między innymi bisfosfoniany, denosumab, romosozumab, tiazyd oraz diuretyki pętlowe, kortykosteroidy układowe oraz lit. U pacjentów leczonych jednocześnie tymi produktami leczniczymi należy obserwować zmiany stężenia wapnia w surowicy.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Ciąża Nie ma danych lub istnieją tylko ograniczone dane dotyczące stosowania produktu leczniczego Yorvipath u kobiet w ciąży. Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na reprodukcję (patrz punkt 5.3). Jednak nie można wykluczyć zagrożenia dla kobiet w ciąży lub rozwijającego się płodu. Decyzja o rozpoczęciu lub odstawieniu leczenia produktem leczniczym Yorvipath w trakcie ciąży powinna uwzględniać stosunek możliwych zagrożeń do korzyści dla kobiet w ciąży. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet w ciąży z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Karmienie piersią Nie wiadomo, czy palopegteryparatyd przenika do mleka ludzkiego. Ponieważ palopegteryparatyd nie jest wchłaniany po podaniu doustnym, jest mało prawdopodobne, aby miał niekorzystny wpływ na dziecko karmione piersią.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWpływ na płodność, ciążę i laktację
Podejmując decyzję o zaprzestaniu karmienia piersią lub leczenia produktem leczniczym Yorvipath, należy wziąć pod uwagę korzyści z karmienia piersią dla dziecka i korzyści z leczenia dla kobiety. Zaleca się monitorowanie stężeń wapnia w surowicy matki u kobiet karmiących piersią z niedoczynnością przytarczyc leczonych produktem leczniczym Yorvipath. Płodność Nie przeprowadzono badań wpływu palopegteryparatydu na płodność człowieka. Dane z badań na zwierzętach nie wskazują, aby podawanie palopegteryparatydu upośledzało płodność (patrz punkt 5.3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Produkt leczniczy Yorvipath nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednak u niektórych pacjentów obserwowano zawroty głowy, stan przedomdleniowy, omdlenia i (lub) niedociśnienie ortostatyczne. Tacy pacjenci powinni powstrzymywać się od prowadzenia pojazdów i obsługiwania maszyn do czasu ustąpienia objawów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa Najczęściej zgłaszanymi działaniami niepożądanymi w badaniach klinicznych z użyciem palopegteryparatydu były reakcje w miejscu wstrzyknięcia (21,6%), ból głowy (18,7%) i parestezja (13,7%). Najpoważniejszą reakcją niepożądaną zgłaszaną w badaniach klinicznych była hiperkalcemia (1,40%). Tabelaryczna lista reakcji niepożądanych Tabela 2 przedstawia reakcje niepożądane u pacjentów leczonych palopegteryparatydem zidentyfikowane we wszystkich badaniach fazy II oraz fazy III przeprowadzonych z grupami kontrolnymi otrzymującymi placebo. Reakcje niepożądane wymienione w poniższej tabeli przedstawiono według klasyfikacji układów i narządów MedDRA oraz częstości występowania zdefiniowanej za pomocą następującej konwencji: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000) i nieznana (częstość występowania nie może być określona na podstawie dostępnych danych).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
W obrębie każdej kategorii częstości występowania reakcje niepożądane przedstawiono według malejącej ciężkości. Tabela 2: Częstość występowania reakcji niepożądanych palopegteryparatydu
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
Klasyfikacja układówi narządów MedDRA Częstość występowania Reakcja niepożądana Zaburzenia metabolizmui odżywiania często hiperkalcemiaa, hipokalcemia Zaburzenia układu nerwowego bardzo często ból głowyd, parestezjaa często zawroty głowya, c, d, omdlenied,stan przedomdleniowyd Zaburzenia serca często palpitacjed, zespół posturalnej tachykardii ortostatycznejd Zaburzenia naczyniowe często niedociśnienie ortostatyczned niezbyt często nadciśnieniee Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia często ból jamy ustnej i gardła Zaburzenia żołądka i jelit bardzo często nudnościa często biegunkaa, zaparcia, wymioty,dyskomfort w jamie brzusznej, ból w jamie brzusznej Zaburzenia skóry i tkanki podskórnej często wysypka, reakcja nadwrażliwości na światło Zaburzenia mięśniowo- szkieletowe i tkanki łącznej często ból stawów, mialgia, drżenie mięśnif, ból mięśniowo-szkieletowyf Zaburzenia nerek i dróg moczowych niezbyt często nokturiae częstość nieznana wielomocze Zaburzenia ogólne i stany w miejscu podania bardzo często reakcje w miejscu wstrzyknięciaa,b, zmęczenie często astenia, pragnienie niezbyt często dyskomfort w klatce piersiowejf, ból w klatce piersiowejf Badania diagnostyczne częstość nieznana gęstość kości zmniejszona - CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
a W przypadku tych reakcji niepożądanych pierwsze wystąpienie pojawiało się niemal wyłącznie w ciągu pierwszych 3 miesięcy leczenia (okres dostosowywania dawki). b Reakcje w miejscu wstrzyknięcia obejmują reakcję w miejscu wstrzyknięcia, rumień w miejscu wstrzyknięcia, zasiniaczenie w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia, krwotok w miejscu wstrzyknięcia, wysypkę w miejscu wstrzyknięcia oraz opuchliznę w miejscu wstrzyknięcia. c Zawroty głowy obejmują zawroty głowy i posturalne zawroty głowy. d Objawy związane z rozszerzeniem naczyń krwionośnych obejmowały posturalne zawroty głowy, ból głowy, palpitacje, zespół posturalnej tachykardii ortostatycznej, niedociśnienie ortostatyczne, obniżone ortostatyczne ciśnienie krwi i omdlenie. Objawy związane z rozszerzeniem naczyń krwionośnych (zidentyfikowane w badaniach klinicznych) występowały częściej podczas pierwszych 3 miesięcy leczenia i stanowiły podzbiór wszystkich zdarzeń zgłaszanych jako reakcje niepożądane.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
W badaniu TCP-304 w ciągu pierwszych 3 miesięcy wystąpiły łącznie 3 zdarzenia (u 2 pacjentów) uznane za związane z palopegteryparatydem: posturalne zawroty głowy (n=1) oraz ból głowy i palpitacje (n=1). e Objawy przedmiotowe i podmiotowe potencjalnie związane z hiperkalcemią, jak zaobserwowano w badaniach klinicznych. f Objawy przedmiotowe i podmiotowe potencjalnie związane z hipokalcemią, jak zaobserwowano w badaniach klinicznych. Opis wybranych reakcji niepożądanych Hiperkalcemia Podczas stosowania produktu leczniczego Yorvipath zgłaszano przypadki hiperkalcemii. Częstość występowania hiperkalcemii była wyższa u pacjentów leczonych produktem leczniczym Yorvipath w porównaniu z placebo. W trakcie okresu zaślepionego objawową hiperkalcemię zgłoszono u 8,6% pacjentów leczonych produktem leczniczym Yorvipath i wszystkie zgłoszone przypadki wystąpiły w ciągu pierwszych 3 miesięcy po rozpoczęciu leczenia produktem leczniczym Yorvipath.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
Immunogenność U pacjentów mogą pojawić się przeciwciała przeciwko palopegteryparatydowi. Odsetek pacjentów z dodatnim wynikiem testu na przeciwciała wiążące był niski w dowolnym momencie leczenia — 0,7% miało niskie miano przeciwciał nieneutralizujących przeciwko PTH, a 5% miało niskie miano związanych z leczeniem przeciwciał przeciwko PEG. U 2,2% pacjentów leczonych palopegteryparatydem z wcześniej nabytymi przeciwciałami przeciwko PEG zaobserwowano przejściowy wpływ na PK (zwiększony klirens całkowitego PTH, mPEG i zmniejszenie stężenia PTH) oraz zmniejszenie stężenia wapnia w surowicy. Jednakże skuteczność terapeutyczna była utrzymana dzięki odpowiedniemu dostosowaniu dawki palopegteryparatydu zgodnie z algorytmem dostosowywania dawki w badaniu. Reakcje w miejscu wstrzyknięcia Reakcje w miejscu wstrzyknięcia były najczęściej zgłaszanymi w badaniach klinicznych reakcjami niepożądanymi (mediana czasu wystąpienia wynosiła 2,5 dnia; częstość występowania 21,6%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
Najczęstszymi reakcjami w miejscu wstrzyknięcia były rumień miejscowy (wszystkie o średnicy < 5 cm, a większość od 0 do < 2 cm) o nasileniu łagodnym lub umiarkowanym (stopień 1 lub 2) z medianą czasu trwania 72 godziny. Wszystkie reakcje w miejscu wstrzyknięcia ustępowały bez leczenia, żadna nie była ciężka ani nie prowadziła do przerwania leczenia. Objawy związane z rozszerzeniem naczyń krwionośnych Podczas stosowania leku Yorvipath zgłaszano objawy rozszerzenia naczyń krwionośnych. Objawy te są zwykle przemijające i ustępują bez leczenia; żaden nie był poważny ani nie doprowadził do przerwania leczenia. W przypadku wystąpienia objawów zaleca się dawkowanie przed snem, w pozycji leżącej. Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDziałania niepożądane
Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzedawkowanie
4.9 Przedawkowanie W razie przedawkowania pacjent powinien być obserwowany przez fachowy personel medyczny. Przedawkowanie może powodować hiperkalcemię, której objawy mogą obejmować odwodnienie, palpitacje serca, zmiany EKG, niedociśnienie, nudności, wymioty, zawroty głowy, osłabienie mięśni oraz splątanie. Pacjent z ciężką hiperkalcemią może wymagać obserwacji i opieki medycznej (patrz punkt 4.4). Jeden przypadek około 3-krotnego przypadkowego przedawkowania przepisanej dawki trwającego ponad 7 kolejnych dni spowodował stężenie wapnia w surowicy wynoszące aż 16,1 mg/dl, pacjent miał objawy i wymagał interwencji medycznej. Po wstrzymaniu podawania palopegteryparatydu, wapnia oraz czynnej witaminy D pacjent wyzdrowiał i ponownie rozpoczął przyjmowanie prawidłowej dawki.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: Hemostaza wapnia, parathormony i analogi, kod ATC: H05AA05 Mechanizm działania Endogenny parathormon (PTH) jest wydzielany przez przytarczyce jako polipeptyd o długości 84 aminokwasów. PTH wywiera działanie za pośrednictwem receptorów parathormonu na powierzchni komórek, na przykład ulegającego ekspresji w tkance kości, nerek i nerwów. Aktywacja PTH1R stymuluje obrót kostny, zwiększa wchłanianie zwrotne wapnia i wydalanie fosforanów w nerkach oraz ułatwia syntezę czynnej witaminy D. Palopegteryparatyd jest prolekiem zawierającym PTH(1-34) sprzężony z nośnikiem glikolu metoksypolietylenowego (mPEG) poprzez zastrzeżony łącznik TransCon. PTH(1-34) i jego główny metabolit, PTH(1-33) mają podobne powinowactwo oraz aktywują PTH1R jak endogenny PTH. W warunkach fizjologicznych PTH jest w kontrolowany sposób odszczepiany od palopegteryparatydu zapewniając ciągłą ekspozycję ogólnoustrojową na aktywny PTH.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Skuteczność kliniczna i bezpieczeństwo stosowania Badanie z udziałem pacjentów z ustaloną niedoczynnością przytarczyc W kluczowym badaniu klinicznym fazy III PaTHway (TCP-304) oceniano skuteczność i bezpieczeństwo stosowania produktu leczniczego Yorvipath jako terapii zastępującej PTH u osób dorosłych z niedoczynnością przytarczyc. 26-tygodniowy, prowadzony metodą podwójnie ślepej próby, kontrolowany placebo okres badania klinicznego obejmował pacjentów randomizowanych (3:1) do grupy przyjmującej produkt leczniczy Yorvipath w dawce początkowej 18 mikrogramów/dobę lub placebo podawane jednocześnie z terapią konwencjonalną (suplement wapnia i czynna witamina D). Randomizacja była stratyfikowana według etiologii niedoczynności przytarczyc (tj. po zabiegu chirurgicznym wobec wszystkich innych przyczyn).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Badane leczenie (palopegteryparatyd lub placebo) i terapia konwencjonalna były następnie dostosowywane według algorytmu dawkowania opartego na stężeniach wapnia w surowicy skorygowanych o stężenie albumin. Średnia wieku pacjentów podczas rekrutacji wynosiła 49 lat (od 19 do 78 lat; 12% miało ≥ 65 lat) i większość pacjentów była kobietami (78%) oraz rasy białej (93%). Osiemdziesiąt pięć procent (85%) miało niedoczynność przytarczyc nabytą po operacji szyi. Spośród pacjentów z innymi etiologiami niedoczynności przytarczyc 7 (8,5%) pacjentów miało chorobę idiopatyczną, 2 miało autoimmunologiczny zespół niedoczynności wielogruczołowej typu 1 (APS-1), 1 miał hipokalcemię autosomalną dominującą typu 1 (ADH1, mutacja CaSR), 1 miał zespół DiGeorge i 1 miał niedoczynności przytarczyc, głuchotę czuciowo-nerwową oraz zespół dysplazji nerek (HDR) (mutacja GATA3).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Przed randomizacją wszyscy pacjenci zostali poddani trwającemu około 4 tygodnie okresowi przesiewowemu, podczas którego suplementy wapnia oraz czynnej witaminy D dostosowano, aby osiągnąć stężenie wapnia w surowicy skorygowane o stężenie albumin wynoszące od 1,95 do 2,64 mmol/l (od 7,8 do 10,6 mg/dl), stężenie magnezu ≥ 0,53 mmol/l (≥ 1,3 mg/dl) i poniżej górnego zakresu referencyjnego normy oraz stężenie 25(OH) witaminy D od 50 do 200 nmol/l (od 20 do 80 ng/ml). W przypadku terapii konwencjonalnej pacjenci byli leczeni średnim wyjściowymi dawkami wapnia (pierwiastkowego) wynoszącymi 1839 mg/dobę. Średnie wyjściowe dawki czynnej witaminy D wynosiły 0,75 mikrograma/dobę u pacjentów leczonych kalcytriolem (n=70) oraz 2,3 mikrograma/dobę u pacjentów leczonych alfakalcydolem (n=12). Średnie wyjściowe stężenie wapnia w surowicy skorygowane o stężenie albumin oraz średnie 24-godzinne stężenie wapnia w moczu były podobne w obu grupach leczenia: średnie stężenie wapnia w surowicy wynosiło 2,2 mmol/l (8,8 mg/dl) oraz 2,15 mmol/l (8,6 mg/dl), a średnie 24-godzinne stężenie wapnia w moczu wynosiło 392 mg/dobę i 329 mg/dobę odpowiednio w przypadku produktu leczniczego Yorvipath oraz placebo.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Pierwszorzędowy punkt końcowy Pierwszorzędowy złożony punkt końcowy był definiowany jako odsetek pacjentów w tygodniu 26, którzy osiągnęli: stężenia wapnia w surowicy w zakresie prawidłowym (od 2,07 do 2,64 mmol/l [od 8,3 do 10,6 mg/dl]), niezależnie od terapii konwencjonalnej definiowanej jako niewymagająca czynnej witaminy D oraz ≤ 600 mg/dobę suplementacji wapnia i bez zwiększenia przepisanego badanego leczenia w ciągu 4 tygodni przed tygodniem 26. Kluczowe drugorzędowe punkty końcowe obejmowały podzbiór punktacji domeny skali doświadczeń pacjentów z niedoczynnością przytarczyc (ang. Hypoparathyroidism Patient Experience Scale, HPES) oraz punktacji podskali skróconego formularza 36-elementowego (ang. 36-Item Short Form Survey, SF-36). Liczbę pacjentów spełniających pierwszorzędowy złożony punkt końcowy w porównaniu z grupą placebo i każdym składnikiem pierwszorzędowego punktu końcowego w tygodniu 26 przedstawiono w tabeli 3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Tabela 3: TCP-304: współczynnik odpowiedzi oparty na pierwszorzędowym punkcie końcowym w tygodniu 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Yorvipath (N=61) (n, %) Placebo (N=21) (n, %) Różnica współczynnikaodpowiedzi (95% CI) Odpowiedź w tygodniu 26 48 (78,7%) 1 (4,8%) 74,0%(60,4%; 87,6%)p < 0,0001 Odpowiedź dla każdego składnika Stężenie wapnia w surowicy skorygowaneo stężenie albumin w zakresie prawidłowyma 49 (80,3%) 10 (47,6%) 32,7%(9,2%; 56,3%) Niezależność od czynnej witaminy Db 60 (98,4%) 5 (23,8%) 74,6%(56,1%; 93,1%) Niezależność od terapeutycznych dawek wapniac 57 (93,4%) 1 (4,8%) 88,7%(77,7%; 99,7%) Brak zwiększenia dawki w grupie produktu leczniczego Yorvipathd 57 (93,4%) 12 (57,1%) 36,4%(14,2%; 58,5%) - CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
a Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosił od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl). b Wszystkie codzienne stałe dawki czynnej witaminy D równe zero ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. c Średnie codzienne stałe dawki pierwiastkowego wapnia ≤ 600 mg ORAZ stosowanie dawek PRN przez ≤ 7 dni w ciągu 4 tygodni przed wizytą po 26 tygodniach. d Brak zwiększenia dawki produktu leczniczego Yorvipath w ciągu 4 tygodni przed wizytą po 26 tygodniach. Skróty: CI: przedział ufności; PRN: pro re nata. Drugorzędowe punkty końcowe Przyjmowanie terapii konwencjonalnej: dawki wapnia i czynnej witaminy D. W badaniu fazy III PaTHway, w tygodniu 26, 93% (57/61) pacjentów w grupie produktu leczniczego Yorvipath było w stanie odstawić terapię konwencjonalną (tj. odstawić czynną witaminę D oraz terapeutyczne dawki wapnia).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Wszyscy pacjenci w grupie produktu leczniczego Yorvipath odstawili czynną witaminę D do tygodnia 8 i utrzymywali zmniejszenie terapeutycznych dawek wapnia. Występowało znaczące zmniejszenie przyjmowania terapii konwencjonalnej w grupie produktu leczniczego Yorvipath od wartości wyjściowej do tygodnia 26 w porównaniu z placebo: czynnej witaminy D (nominalna wartość p < 0,0001), dawki wapnia (nominalna wartość p = 0,0003) oraz codziennego obciążenia tabletkami (nominalna wartość p < 0,0001) (tabela 4). Tabela 4: Drugorzędowe punkty końcowe: przyjmowanie terapii konwencjonalnej w tygodniu 26 — okres zaślepiony (populacja ITT)
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Yorvipath (n/N=60/61)a Placebo (n/N=19/21)a Nominalnawartość p Wartość początkowa Tydzień 26 Wartość początkowa Tydzień 26 Suplementacyjna dawkaczynnej witaminy D (µg), średnia (SD) 1,0 (0,7) 0,0 (0,0) 1,0 (0,6) 0,6 (0,7) < 0,0001 Suplementacyjna dawka wapnia (mg), średnia (SD) 1737 (907) 274 (177) 2089 (1448) 1847 (1326) 0,0003 Codzienne obciążenie tabletkami (liczba tabletekterapii konwencjonalnej), średnia (SD) 6,6 (2,1) 0,5 (1,7) 6,3 (2,8) 5,4 (3,2) < 0,0001 - CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Nominalna wartość p z badań różnic zmiany od wartości wyjściowej do tygodnia 26 między produktem leczniczym Yorvipath oraz placebo. a N to liczba pacjentów w populacji ITT; n to liczba pacjentów z danymi zarówno wyjściowymi, jak i w tygodniu 26. Parametry biochemiczne surowicy Średnia wartość wapnia w surowicy początkowo wzrosła i utrzymywała się w zakresie prawidłowym u pacjentów leczonych palopegteryparatydem (rysunek 2). U pacjentów przyjmujących placebo stężenia wapnia w surowicy nieco zmniejszyły się, spadając poniżej zakresu prawidłowego w tygodniu 2 (średnia obserwowana wartość: 2,06 mmol/l) i w tygodniu 26 (średnia obserwowana wartość: 2,06 mmol/l). Różnica średniej najmniejszych kwadratów leczenia między produktem leczniczym Yorvipath i placebo wynosiła 0,17 mmol/l (95% CI: 0,100; 0,247; nominalna wartość p < 0,0001) w tygodniu 26. Rysunek 2: Wapń w surowicy (średnia ±SE) według wizyty — okres zaślepiony (populacja ITT) 3 2,5 2 1,5 0 2 4 6 8 10 12 14 16 18 20 22 24 26
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Średnie (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mmol/l) Czas (tygodnie)
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Palopegteryparatyd
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Placebo Średnie stężenia fosforanu w surowicy u pacjentów leczonych palopegteryparatydem były w zakresie prawidłowym wyjściowo i utrzymywały się w zakresie prawidłowym do tygodnia 26 (średnia zmiana od wartości wyjściowej do tygodnia 26 wynosiła -0,13 mmol/l). Średni iloczyn wapnia i fosforanów w surowicy zmniejszył się u pacjentów leczonych produktem leczniczym Yorvipath i pozostawał stabilny w zakresie prawidłowym do tygodnia 26. 24-godzinne wydalanie wapnia w moczu Terapia produktem leczniczym Yorvipath normalizowała 24-godzinną średnią wydalania wapnia w moczu i wykazywała większe zmniejszenie 24-godzinnego stężenia wapnia w moczu niż placebo. Dzieci i młodzież Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań produktu leczniczego Yorvipath w jednej lub kilku podgrupach populacji dzieci i młodzieży z niedoczynnością przytarczyc, zgodnie z warunkami zawartymi w decyzji dotyczącej planu badań dzieci i młodzieży (PIP, ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakodynamiczne
Paediatric Investigation Plan) we wskazaniu leczenia niedoczynności przytarczyc.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Wchłanianie Po codziennym podawaniu podskórnym palopegteryparatyd uwalnia PTH za pośrednictwem autoodszczepienia łącznika TransCon podlegając kinetyce pierwszorzędowej, dając ciągłą ekspozycję przez 24 godziny w szacowanym zakresie prawidłowym (rysunek 3). Rysunek 3: Średni uwalniany PTH* po podawaniu podskórnym palopegteryparatydu w stanie stacjonalnym u pacjentów z niedoczynnością przytarczyc 40 30 20 10 0 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
Średni (+/-SE) uwalniany PTH (pg/ml) Czas (godz.) Szacowany zakres prawidłowy dla PTH(1-34) wynosi około 4 do 26 pg/ml. Jest on obliczony na podstawie PTH(1-34) stanowiącego 40% masy cząsteczkowej PTH(1-84)** i zakresu prawidłowego (10 do 65 pg/ml) dla PTH(1-84). * Średnia dawka palopegteryparatydu (zakres): 22,3 (12-33) µg/dobę, n=7. uwolniony PTH: suma PTH(1-34) i PTH(1-33). ** PTH(1-84) = endogenny parathormon. U pacjentów z niedoczynnością przytarczyc podawanie palopegteryparatydu odpowiadało 18 µg PTH(1-34)/dobę, przewidywane maksymalne stężenie w osoczu (C max ) (CV%) palopegteryparatydu wynosiło 5,18 ng/ml (36%), a przewidywana wartość C max (CV%) dla uwalnianego PTH wynosiła 6,9 pg/ml (22%) z medianą czasu do osiągnięcia maksymalnych stężeń (T max ) wynoszącą 4 godziny. Przewidywana ekspozycja w ciągu 24-godzinnego odstępu między dawkami (pole pod krzywą, ang. area under the curve, AUC) (CV%) dla uwalnianego PTH wynosiła 150 pg*h/ml (22%).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
Po podaniu wielu podskórnych dawek palopegteryparatydu w zakresie od 12 do 24 µg PTH(1-34)/dobę stężenia palopegteryparatydu i uwalnianego PTH rosły w sposób proporcjonalny do dawki, osiągając stan stacjonarny w ciągu odpowiednio około 10 i 7 dni. Stosunek wartości maksymalnej do minimalnej był niski, wynosił około 1,1 oraz 1,5 przez 24 godziny stanu stacjonarnego odpowiednio dla palopegteryparatydu i uwalnianego PTH. Palopegteryparatyd ulegał akumulacji po wielu dawkach do 18-krotności wartości AUC. Dystrybucja Pozorną objętość dystrybucji (CV%) palopegteryparatydu szacuje się na 4,8 l (50%) oraz do 8,7 l (18%) dla uwalnianego PTH. Metabolizm PTH uwalniany z palopegteryparatydu składa się z PTH(1-34) i metabolitu PTH(1-33). PTH jest metabolizowany i usuwany w nerkach. Eliminacja U zdrowych osób dorosłych klirens (CV%) palopegteryparatydu w stanie stacjonarnym szacuje się na 0,58 l/dobę (52%) z przewidywanym okresem półtrwania wynoszącym 70 godzin.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
Pozorny okres półtrwania PTH uwalnianego z palopegteryparatydu wynosi około 60 godzin. W wątrobie większość PTH jest rozszczepiana przez katepsyny. W nerkach niewielka ilość PTH wiąże się z PTH1R, ale większość jest wydalana poprzez filtrację kłębuszkową. Zależności farmakokinetyczno-farmakodynamiczne W cząstkowym badaniu farmakodynamiki/farmakokinetyki u pacjentów z niedoczynnością przytarczyc, codzienne podskórne podawanie palopegteryparatydu (średnia dawka (zakres): 22,3 (12– 33) µg PTH (1-34)dobę) zwiększała stężenie wapnia w surowicy do zakresu prawidłowego (patrz rysunek 4). Zwiększenie stężeń wapnia w surowicy następowało w sposób zależny od dawki wspierając możliwość dostosowania dawki palopegteryparatydu odpowiednio do zmierzonych wartości wapnia w surowicy u danego pacjenta. mmol/l Rysunek 4: Średnie stężenia wapnia w surowicy skorygowane o stężenie albumin po podawaniu podskórnym palopegteryparatydu w stanie stacjonarnym u pacjentów z niedoczynnością przytarczyc 12 11 10 9 8 7 3 2,75 2,5 2,25 2 1,75 0 4 8 12 16 20 24
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
Średnia (+/-SE) stężenie wapnia w surowicy skorygowane o stężenie albumin (mg/dl) Czas (godz.) Zakres prawidłowy stężenia wapnia w surowicy skorygowanego o stężenie albumin wynosi od 2,07 do 2,64 mmol/l (od 8,3 do 10,6 mg/dl), co oznaczono szarym cieniowaniem. Średnia dawka palopegteryparatydu (zakres): 22,3 (12–33) µg PTH(1- 34)/dobę, n=7. Szczególne grupy pacjentów Na farmakokinetykę uwalnianego PTH nie wpływa płeć ani masa ciała. Dane dotyczące rasy i pochodzenia etnicznego nie wykazują żadnych trendów wskazujących na różnice, ale dostępne dane są zbyt ograniczone do postawienia rozstrzygających wniosków. Osoby w starszym wieku Na farmakokinetykę uwalnianego PTH nie wpływa wiek (od 19 do 76 lat). Zaburzenia czynności nerek Produkt leczniczy Yorvipath podawano pacjentom z niedoczynnością przytarczyc z eGFR ≥ 30 ml/min w długotrwałych badaniach klinicznych bez konieczności dostosowywania dawki poza algorytmem dostosowywania dawki w ramach badania.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlWłaściwości farmakokinetyczne
Nie przeprowadzono badań klinicznych z udziałem pacjentów z niedoczynnością przytarczyc oraz zaburzeniem czynności nerek (< 30 ml/min) lub dializowanych. W badaniu, w którym produkt leczniczy Yorvipath podawano jako pojedynczą dawkę uczestnikom bez niedoczynności przytarczyc, ekspozycja na palopegteryparatyd i uzyskiwane stężenia wapnia w surowicy były podobne jak u uczestników z łagodnym, umiarkowanym lub ciężkim upośledzeniem czynności nerek w porównaniu do pacjentów bez zaburzeń czynności nerek.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie Nie ujawniono szczególnego zagrożenia dla człowieka wynikającego z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, genotoksyczności i tolerancji miejscowej przeprowadzonych na palopegteryparatydzie. U zwierząt wszystkich zastosowanych gatunków podanie wielokrotne największych dawek powodowało niepożądaną uporczywą hiperkalcemię, która w niektórych badaniach prowadziła do przedwczesnego zgonu/eutanazji, objawów klinicznych, utrata masy ciała i (lub) mineralizacji tkanki miękkiej obserwowanej głównie w nerkach. Wyniki te są uważane za skutki nieustannie przesadnej farmakologii PTH i nie mają one znaczenia w warunkach klinicznych, gdzie dawkę dostosowuje się w celu zapewnienia normalizacji stężenia wapnia w surowicy Zgodnie z oczekiwanymi skutkami farmakologicznymi, powtarzane codzienne podawanie palopegteryparatydu zwiększało obrót kostny u szczurów.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzedkliniczne dane o bezpieczeństwie
Przy małych dawkach (2-krotność maksymalnej zalecanej dawki dla człowieka, MRHD) na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny indukował netto całkowite kataboliczne skutki kostne. Przy dużych dawkach (5-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów, zwiększony obrót kostny powodował netto anaboliczny skutek kostny. Dysplazja nasad kości była obserwowana przy najwyższej dawce (9-krotność MRHD na podstawie ekspozycji na PTH według AUC) u szczurów. Skutki te nie mają znaczenia w warunkach klinicznych, w których dawki leku Yorvipath dostosowuje się indywidualnie. Nie było skutków sercowo-naczyniowych u małp do najwyższej badanej dawki (włącznie) w badaniach dawki pojedynczej (3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ) lub dawki powtarzanej (0,98-krotność MRHD na podstawie ekspozycji na uwalniany PTH według C max ).
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzedkliniczne dane o bezpieczeństwie
Nasilone występowanie kostniakomięsaków obserwowano w badaniach rakotwórczości z krótko żyjącymi analogami PTH u szczurów, ale bez dowodów zwiększonego ryzyka kostniakomięsaka u pacjentów leczonych krótko żyjącymi analogami PTH.Dlatego też nie przeprowadzono żadnego badania nad rakotwórczością palopegteryparatydu. W badaniach reprodukcji na zwierzętach podawanie palopegteryparatydu ciężarnym szczurom i królikom w okresie organogenezy nie dawało dowodów na letalność zarodka, toksyczność dla płodu lub dysmorfogenezę w przypadku podania najwyższych badanych dawek (włącznie) (odpowiednio 8- i 7-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC). Skutki przesadnej farmakologii PTH obserwowano przy najwyższych badanych dawkach u ciężarnych szczurów oraz królików (podwyższone stężenia wapnia w surowicy, zmniejszona masa ciała, zmniejszone spożycie pokarmu i (lub) oznaki kliniczne). Ekspozycje przy poziomie dawkowania, przy którym nie obserwuje się szkodliwych zmian (ang.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlPrzedkliniczne dane o bezpieczeństwie
no observed adverse effect level, NOAEL) w przypadku toksyczności dla matki wynosił 2- i 3-krotność MRHD na podstawie ekspozycji na uwalniany PTH według AUC u odpowiednio ciężarnych szczurów i królików. Nie przeprowadzono badania rozwoju pre- i postnatalnego związanego z palopegteryparatydem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Kwas bursztynowy Mannitol Metakrezol Sodu wodorotlenek Kwas chlorowodorowy (do regulacji pH) Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 3 lata. Po pierwszym otwarciu Przechowywać w temperaturze poniżej 30 °C. Pozostawić zatyczkę wstrzykiwacza na fabrycznie napełnionym wstrzykiwaczu w celu ochrony przed światłem. Produkt leczniczy Yorvipath należy wyrzucić po 14 dniach. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2 °C – 8 °C). Nie zamrażać. Przechowywać w oryginalnym opakowaniu z założoną zatyczką wstrzykiwacza w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po pierwszym otwarciu, patrz punkt 6.3.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDane farmaceutyczne
6.5 Rodzaj i zawartość opakowania Wkład (szkło typu 1) z tłokiem (halobutyl) i laminowanym arkuszem gumowym (halobutyl/izopren) umieszczony w wielodawkowym jednorazowym fabrycznie napełnionym wstrzykiwaczu wykonanym z polipropylenu. Opakowania dwóch fabrycznie napełnionych wstrzykiwaczy oraz 30 igieł jednorazowych na 28 dni leczenia (zapakowane razem w wewnętrznych pudełkach). Każde wewnętrzne pudełko zawiera jeden fabrycznie napełniony wstrzykiwacz oraz 15 igieł na 14 dni leczenia. Yorvipath 168 mikrogramów/0,56 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 168 mikrogramom PTH(1-34) w 0,56 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 6, 9 lub 12 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest niebieski.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDane farmaceutyczne
Yorvipath 294 mikrogramy/0,98 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 294 mikrogramom PTH(1-34) w 0,98 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 15, 18 lub 21 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest pomarańczowy. Yorvipath 420 mikrogramy/1,4 ml roztwór do wstrzykiwań w fabrycznie napełnionym wstrzykiwaczu Każdy fabrycznie napełniony wstrzykiwacz zawiera palopegteryparatyd równoważny 420 mikrogramom PTH(1-34) w 1,4 ml rozpuszczalnika. Fabrycznie napełniony wstrzykiwacz dostarcza dawki 24, 27 lub 30 mikrogramów Kolor mocy na pudełku zewnętrznym, etykiecie wstrzykiwacza i przycisku jest bordowy. 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Przygotowanie dawki Nowy wstrzykiwacz produktu leczniczego Yorvipath należy wyjąć z lodówki na 20 minut przed pierwszym otwarciem.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDane farmaceutyczne
Roztwór powinien być przezroczysty, bezbarwny i wolny od cząstek stałych. Nie należy wstrzykiwać produktu leczniczego, jeżeli jest mętny lub zawiera cząstki stałe. Każdy fabrycznie napełniony wstrzykiwacz jest przeznaczony do użytku przez jednego pacjenta. Fabrycznie napełniony wstrzykiwacz nie powinien być współdzielony przez pacjentów, nawet jeżeli igła jest zmieniana. Jeżeli fabrycznie napełniony wstrzykiwacz był zamrażany lub narażony na ciepło, należy go wyrzucić. Za każdym razem, gdy fabrycznie napełniony wstrzykiwacz jest przygotowywany do podawania, należy założyć nową igłę. Igieł nie wolno używać ponownie. Może to zapobiec zablokowaniu igieł, zanieczyszczeniu, zakażeniu, wyciekowi roztworu lub niedokładnemu dawkowaniu. Igła iniekcyjna powinna być zdejmowana po każdym wstrzyknięciu, a wstrzykiwacz należy przechowywać bez przymocowanej igły. Igłę należy wyrzucać po każdym wstrzyknięciu.
- CHPL leku Yorvipath, roztwór do wstrzykiwań, 420 mcg/1,4 mlDane farmaceutyczne
Instrukcje przygotowywania i podawania produktu leczniczego Yorvipath podano w ulotce dołączonej do opakowania oraz instrukcji użycia. Utylizacja Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO Eladynos 80 mikrogramów/dawkę roztwór do wstrzykiwań we wstrzykiwaczu. 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY Każda dawka (40 mikrolitrów) zawiera 80 mikrogramów abaloparatydu. Każdy wstrzykiwacz półautomatyczny napełniony zawiera 3 mg abaloparatydu w 1,5 mlroztworu (co odpowiada 2 miligramom na ml). Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (płyn do wstrzykiwań). Bezbarwny, przejrzysty roztwór.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWskazania do stosowania
4.1 Wskazania do stosowania Leczenie osteoporozy u kobiet w okresie pomenopauzalnym, u których występuje zwiększone ryzyko złamań (patrz punkt 5.1).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDawkowanie
4.2 Dawkowanie i sposób podawania Dawkowanie Zalecana dawka to 80 mikrogramów raz na dobę. Maksymalny całkowity czas trwania leczenia abaloparatydem powinien wynosić 18 miesięcy (patrz punkty 4.4 i 5.1). Pacjentki powinny otrzymywać suplementację wapnia i witaminy D, jeśli ich zawartość w diecie jest niewystarczająca. Po zakończeniu leczenia abaloparatydem pacjentki mogą kontynuować leczenie osteoporozy innymi lekami, takimi jak bifosfoniany. Pominięcie dawki Jeśli pacjentka zapomni lub nie może podać sobie dawki o zwykłej porze, może ją wstrzyknąć w ciągu 12 godzin od zwykłej pory podawania produktu leczniczego. Nie należy podawać więcej niż jednego wstrzyknięcia tego samego dnia i nie należy próbować uzupełniać pominiętej dawki. Szczególne grupy pacjentek Pacjentki w podeszłym wieku Nie jest wymagana modyfikacja dawki w zależności od wieku (patrz punkt 5.2).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDawkowanie
Zaburzenia czynności nerek Nie wolno stosować abaloparatydu u pacjentek z ciężkimi zaburzeniami czynności nerek, w tym pacjentek ze schyłkową niewydolnością nerek (patrz punkt 4.3). U pacjentek z łagodnymi do umiarkowanych zaburzeniami czynności nerek modyfikacja dawki nie jest wymagana (patrz punkt 5.2). Zaburzenia czynności wątroby Dane dotyczące pacjentek z zaburzeniami czynności wątroby nie są dostępne. U tych pacjentek modyfikacja dawki nie jest wymagana, ponieważ jest mało prawdopodobne, aby zaburzenia czynności wątroby miały znaczący wpływ na ekspozycję na abaloparatyd (patrz punkt 5.2). Dzieci i młodzież Abaloparatydu nie należy stosować u dzieci i młodzieży w wieku poniżej 18 lat ze względu na bezpieczeństwo stosowania (patrz punkt 5.3). Sposób podawania Wyłącznie do podawania podskórnego. Pierwsze wstrzyknięcie (wstrzyknięcia) pacjentka lub opiekun powinni wykonać pod nadzorem odpowiednio wykwalifikowanej osoby należącej do fachowego personelu medycznego (patrz punkt 4.4).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDawkowanie
Pacjentki i (lub) opiekunów należy przeszkolić w zakresie podskórnego podawania abaloparatydu (patrz punkt 6.6). Do każdego opakowania dołączono szczegółową instrukcję dotyczącą prawidłowego stosowania wstrzykiwacza. Abaloparatyd należy wstrzykiwać w okolicę podbrzusza. Miejsce wstrzyknięcia należy zmieniać codziennie. Wstrzyknięcia należy podawać codziennie, w przybliżeniu o tej samej porze.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1. - Ciąża i karmienie piersią (patrz punkt 4.6). - Kobiety w wieku rozrodczym (patrz punkty 4.6 i 5.3). - Istniejąca wcześniej hiperkalcemia. - Ciężkie zaburzenia czynności nerek (patrz punkty 4.2 i 5.2). - Niewyjaśnione zwiększenie aktywności fosfatazy alkalicznej. - Pacjentki ze stwierdzonymi czynnikami ryzyka wystąpienia kostniakomięsaka, jak np. pacjentki, które poddawano wcześniej radioterapii wiązką zewnętrzną lub brachyterapii kośćca (patrz punkt 5.3). - Pacjentki z nowotworami złośliwymi kośćca lub przerzutami do kości.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Niedociśnienie ortostatyczne i zwiększenie częstości akcji serca Podczas leczenia abaloparatydem może wystąpić niedociśnienie ortostatyczne i przemijające zwiększenie częstości akcji serca, zazwyczaj w ciągu 4 godzin od wstrzyknięcia. Objawy mogą obejmować zawroty głowy, kołatanie serca, częstoskurcz lub nudności i mogą ustąpić po zmianie pozycji na leżącą. Pierwsze wstrzyknięcie (wstrzyknięcia) abaloparatydu należy wykonać pod nadzorem odpowiednio wykwalifikowanej osoby należącej do fachowego personelu medycznego, która może obserwować stan pacjentki przez pierwszą godzinę od wstrzyknięcia. Abaloparatyd należy zawsze podawać w miejscu, w którym pacjent może w razie konieczności usiąść lub położyć się. Abaloparatyd może mieć działanie rozszerzające na mięśnie gładkie naczyń i dodatnie działanie chronotropowe/inotropowe na mięsień sercowy. Istotna jest indywidualna ocena stosunku korzyści do ryzyka.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlSpecjalne środki ostrozności
Przed rozpoczęciem leczenia abaloparatydem należy ocenić ciśnienie krwi, stan kardiologiczny i EKG. Pacjentki z chorobą serca należy monitorować, gdyż choroba ta może się zaostrzyć. W przypadku wystąpienia niedociśnienia ortostatycznego lub ciężkich objawów ze strony układu sercowo-naczyniowego należy przerwać leczenie. Hiperkalcemia Po wstrzyknięciu abaloparatydu pacjentkom z prawidłowym stężeniem wapnia obserwowano przemijające zwiększenie stężenia wapnia w surowicy. Maksymalne stężenie wapnia w surowicy występowało po około 4 godzinach i powracało do wartości początkowej po 24 godzinach od podania każdej dawki. Z tego powodu próbki krwi w celu oznaczenia stężenia wapnia w surowicy należy pobierać po około 24 godzinach od wykonania ostatniego wstrzyknięcia. U pacjentek, u których nie występują dodatkowe czynniki ryzyka hiperkalcemii, rutynowe monitorowanie stężenia wapnia nie jest wymagane. Hiperkalciuria i kamica moczowa Abaloparatyd może powodować hiperkalciurię.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlSpecjalne środki ostrozności
Nie wiadomo, czy abaloparatyd może nasilać kamicę moczową u pacjentek z czynną kamicą moczową lub kamicą moczową w wywiadzie. W przypadku podejrzewania czynnej kamicy moczowej lub istniejącej wcześniej hiperkalciurii należy rozważyć pomiar wydalania wapnia z moczem. Długość leczenia Maksymalna całkowita długość leczenia abaloparatydem powinna wynosić 18 miesięcy. W badaniach na szczurach wykazano zwiększoną częstość występowania kostniakomięsaków w przypadku długotrwałego podawania abaloparatydu (patrz punkt 5.3). Zawartość sodu Produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji Nie przeprowadzono dedykowanych badań dotyczących interakcji typu lek-lek z abaloparatydem. Biorąc pod uwagę właściwości farmakokinetyczne abaloparatydu prawdopodobieństwo wystąpienia potencjalnych interakcji uważa się za niewielkie. Nie ma danych dotyczących skuteczności abaloparatydu u pacjentek leczonych wcześniej lub jednocześnie bisfosfonianami lub glukokortykoidami. Jednoczesne stosowanie wazoaktywnych produktów leczniczych może predysponować do występowania niedociśnienia ortostatycznego, ponieważ działanie abaloparatydu obniżające ciśnienie krwi może ulec nasileniu, patrz punkt 4.4. Sporadyczne opisy przypadków sugerują, że hiperkalcemia może predysponować do wystąpienia działania toksycznego glikozydów naparstnicy. Ponieważ wykazano, że abaloparatyd zwiększa stężenie wapnia w surowicy, należy zachować ostrożność w przypadku stosowania u pacjentek przyjmujących glikozydy naparstnicy.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Ten produkt leczniczy nie jest wskazany do stosowania u kobiet w wieku rozrodczym. Nie należy stosować go u kobiet, które są lub mogą być w ciąży, lub karmiących piersią (patrz punkty 4.1 i 4.3). Ciąża Produkt leczniczy Eladynos jest przeciwwskazany do stosowania w okresie ciąży (patrz punkt 4.3). Karmienie piersią Nie wiadomo, czy abaloparatyd przenika do mleka ludzkiego. Nie można wykluczyć zagrożenia dla noworodków/dzieci. Produkt leczniczy Eladynos jest przeciwwskazany podczas karmienia piersią (patrz punkt 4.3). Płodność Dane dotyczące wpływu abaloparatydu na płodność u ludzi nie są dostępne. Badania na szczurach z zastosowaniem abaloparatydu nie wykazały wpływu na płodność u samców (patrz punkt 5.3).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Abaloparatyd nie wpływa lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Po podaniu abaloparatydu może wystąpić przemijające niedociśnienie ortostatyczne lub zawroty głowy (patrz punkt 4.8). Pacjentki te nie powinny prowadzić pojazdów mechanicznych ani obsługiwać maszyn do czasu ustąpienia objawów.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
4.8 Działania niepożądane Podsumowanie profilu bezpieczeństwa stosowania Najczęściej zgłaszanymi objawami niepożądanymi u pacjentek leczonych abaloparatydem w badaniu ACTIVE były hiperkalciuria (15,6%), zawroty głowy (11,1%), ból pleców (8,6%), nudności (8,5%), ból głowy (8,5%), ból stawów (8,4%), nadciśnienie tętnicze (6,8%), reakcja w miejscu wstrzyknięcia (6,2%) i kołatanie serca (5,6%). Tabelaryczne zestawienie działań niepożądanych Spośród pacjentek w badaniu ACTIVE z zastosowaniem abaloparatydu, 90,3% pacjentek otrzymujących abaloparatyd i 88,4% pacjentek otrzymujących placebo zgłaszało co najmniej 1 zdarzenie niepożądane. Działania niepożądane związane ze stosowaniem abaloparatydu zgłaszane u pacjentek z osteoporozą w badaniu ACTIVE i w okresie po wprowadzeniu do obrotu podsumowano w tabeli poniżej.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Działania niepożądane sklasyfikowano według następującej konwencji MedDRA: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1 000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1 000), bardzo rzadko (< 1/10 000) i częstość nieznana (częstość nie może być określona na podstawie dostępnych danych). Tabela 1 – Tabelaryczne zestawienie działań niepożądanych
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Zaburzenia układu immunologicznegoNiezbyt często: nadwrażliwośćCzęstość nieznana: reakcja anafilaktyczna Zaburzenia metabolizmu i odżywianiaCzęsto: hiperkalcemia, hiperurikemia Zaburzenia psychiczneCzęsto: bezsenność Zaburzenia układu nerwowegoBardzo często: zawroty głowyCzęsto: ból głowy Zaburzenia sercaCzęsto: kołatanie serca, częstoskurcz Zaburzenia naczynioweCzęsto: nadciśnienie tętniczeNiezbyt często: niedociśnienie ortostatyczne Zaburzenia żołądka i jelitCzęsto: nudności, ból brzucha, zaparcie, biegunka, wymiotyNiezbyt często: wzdęcie brzucha Zaburzenia skóry i tkanki podskórnejCzęsto: świąd, wysypka Zaburzenia mięśniowo-szkieletowe i tkanki łącznejCzęsto: ból pleców, ból stawów, ból kończyn, kurcze mięśni (pleców i kończyn dolnych), ból kości Zaburzenia nerek i dróg moczowychBardzo często: hiperkalciuriaCzęsto: kamica nerkowa Zaburzenia ogólne i stany w miejscu podaniaCzęsto: reakcja w miejscu wstrzyknięcia, zmęczenie, astenia, złe samopoczucieNiezbyt często: ból - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Opis wybranych działań niepożądanych Zwiększenie częstości akcji serca W badaniu odstępu QT średnie zwiększenie częstości akcji serca skorygowane względem placebo wynosiło 14,5 uderzeń na minutę po 15 minutach od podania. To zwiększenie częstości akcji serca było bardziej widoczne w ciągu pierwszej godziny po podaniu dawki i u niektórych uczestniczek obserwowano je do 6 godzin. W badaniu ACTIVE częstość akcji serca mierzono godzinę po podaniu dawki na każdej wizycie związanej z badaniem, przy czym obserwowano medianę zwiększenia częstości akcji serca względem wartości sprzed podania dawki wynoszącą 14 uderzeń na minutę u pacjentek leczonych abaloparatydem w porównaniu z 7 uderzeniami na minutę u pacjentek leczonych placebo. U pacjentek, u których wystąpiło zwiększenie częstości akcji serca o > 20 uderzeń na minutę po 1 godzinie od podania pierwszej dawki, bardziej prawdopodobne było wystąpienie kołatania serca i (lub) zwiększenie częstości akcji serca o > 20 uderzeń na minutę podczas podawania kolejnego wstrzyknięcia.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Działania niepożądane w postaci częstoskurczu i częstoskurczu zatokowego zgłaszano u 1,6% pacjentek otrzymujących abaloparatyd i u 0,4% pacjentek otrzymujących placebo. Niedociśnienie ortostatyczne U kobiet z osteoporozą pomenopauzalną zgłaszano działania niepożądane w postaci niedociśnienia ortostatycznego u 1% pacjentek otrzymujących abaloparatyd i 0,6% pacjentek z grupy placebo. Reakcje w miejscu wstrzyknięcia Abaloparatyd może powodować reakcje w miejscu wstrzyknięcia, w tym zasinienie, rumień, krwotok, nadwrażliwość, ból, wysypka i obrzęk w miejscu wstrzyknięcia. Całkowita częstość występowania w grupie otrzymującej abaloparatyd wynosiła 5,3% w porównaniu z 4,0% w grupie otrzymującej placebo. Wyniki badań laboratoryjnych Stężenie wapnia w surowicy Abaloparatyd może powodować przemijające zwiększenie stężenia wapnia w surowicy oznaczonego po 4 godzinach od podania dawki.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Całkowita częstość występowania hiperkalcemii, zdefiniowanej jako stężenie wapnia w surowicy skorygowane o stężenie albumin ≥ 2,67 mmol/l (lub ≥ 10,7 mg/dl), w grupie otrzymującej abaloparatyd była większa (3,3%) w porównaniu z grupą otrzymującą placebo (0,4%). Kwas moczowy w surowicy Abaloparatyd zwiększał stężenia kwasu moczowego w surowicy. W badaniu ACTIVE u 25% pacjentek w grupie otrzymującej abaloparatyd występowało prawidłowe stężenie kwasu moczowego w punkcie początkowym, które po rozpoczęciu leczenia uległo zwiększeniu powyżej zakresu wartości prawidłowych, w porównaniu z 5% w grupie placebo. Hiperkalciuria i kamica moczowa W badaniu klinicznym z udziałem kobiet z osteoporozą pomenopauzalną całkowita częstość występowania współczynnika wapń: kreatynina w moczu > 0,00113 mmol/µmol (lub > 400 mg/g) był większy w grupie otrzymującej abaloparatyd niż w grupie otrzymującej placebo (odpowiednio 20% w porównaniu z 15%).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Kamicę moczową zgłaszano u 1,4% pacjentek w grupie otrzymującej abaloparatyd i 1,2% pacjentek w grupie otrzymującej placebo. Immunogenność Spośród pacjentek otrzymujących abaloparatyd przez 18 miesięcy, 42,9% wytworzyło przeciwciała przeciwko abaloparatydowi, a u 28,5% wytworzyło w warunkach in vitro przeciwciała neutralizujące. Powstawanie przeciwciał przeciwko abaloparatydowi wiąże się ze zwiększeniem klirensu abaloparatydu. Te zmiany klirensu mogą być związane z obecnością przeciwciał przeciwko abaloparatydowi utrudniającymi dokładne oznaczenie stężenia abaloparatydu w osoczu. Nie obserwowano istotnych klinicznie różnic w bezpieczeństwie stosowania lub skuteczności między pacjentkami, które wytworzyły przeciwciała lub wytworzyły w warunkach in vitro przeciwciała neutralizujące, a pacjentkami, które nie wytworzyły przeciwciał. Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDziałania niepożądane
Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzedawkowanie
4.9 Przedawkowanie Objawy przedmiotowe i podmiotowe W badaniach klinicznych abaloparatyd był podawany podskórnie w dawkach pojedynczych do 320 mikrogramów i w dawkach wielokrotnych do 120 mikrogramów/dobę przez 7 dni. Głównym działaniem niepożądanym ograniczającym dawkę były zawroty głowy przy zmianie pozycji ciała. Objawy przedawkowania, których można się spodziewać, obejmują przemijającą hiperkalciurię, hiperkalcemię, nudności, wymioty, zawroty głowy, kołatanie serca, niedociśnienie ortostatyczne i ból głowy. W programie klinicznym z zastosowaniem wcześniejszej wersji wstrzykiwacza zgłoszono przypadkowe przedawkowanie u pacjentki, która otrzymała 400 mikrogramów w ciągu jednej doby (5-krotność zalecanej dawki klinicznej). U pacjentki wystąpiły astenia, ból głowy, nudności i zawroty głowy pochodzenia błędnikowego. W dniu przedawkowania nie oceniano stężenia wapnia w surowicy, niemniej następnego dnia stężenie wapnia w surowicy pacjentki mieściło się w zakresie wartości prawidłowych.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzedawkowanie
Leczenie przedawkowania Nie ma specyficznej odtrutki na abaloparatyd. Leczenie podejrzewanego przedawkowania może obejmować tymczasowe przerwanie leczenia, monitorowanie stężenia wapnia w surowicy i wdrożenie odpowiednich środków podtrzymujących, takich jak nawadnianie.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: Leki wpływające na homeostazę wapnia, hormony przytarczyc i ich analogi; kod ATC: H05AA04 Mechanizm działania Abaloparatyd to peptyd składający się z 34 aminokwasów, który jest w 41% podobny do hormonu przytarczyc [PTH(1-34)] i w 76% podobny do peptydu PTH-podobnego [PTHrP(1-34)], i jest aktywatorem szlaku sygnałowego receptora PTH1. Abaloparatyd stymuluje tworzenie nowej tkanki kostnej na powierzchni warstwy beleczkowej i korowej poprzez stymulację aktywności osteoblastów. Abaloparatyd powoduje przemijające i ograniczone zwiększenie resorpcji tkanki kostnej i zwiększa gęstość kości. Skuteczność kliniczna i bezpieczeństwo stosowania Skuteczność i bezpieczeństwo stosowania abaloparatydu podawanego raz na dobę oceniano w wieloośrodkowym, prowadzonym metodą podwójnie ślepej próby badaniu klinicznym z randomizacją oraz grupą kontrolną placebo i otwartą grupą kontrolną przyjmującą aktywny lek porównawczy (teryparatyd) (badanie ACTIVE) przez 18 miesięcy leczenia i 1 miesiąc obserwacji z udziałem 2 070 kobiet w okresie pomenopauzalnym w wieku od 50 do 86 lat (średnia wieku 69; 15% było w wieku < 65 lat; 65% było w wieku od 65 do ˂75 lat, a 20% było w wieku ≥ 75 lat), które włączono i zrandomizowano do otrzymywania 80 mikrogramów abaloparatydu (N = 696), placebo (N = 688) lub 20 mikrogramów teryparatydu (N = 686).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Około 76% pacjentek było rasy kaukaskiej, 19% rasy azjatyckiej, a 4% rasy czarnej. Spośród całej populacji badania, 28% było Latynosami. Kobiety przyjmowały codziennie suplementację wapnia (od 500 do 1 000 mg na dobę) i witaminy D (od 400 do 800 j.m. na dobę). Pierwszorzędowym punktem końcowym w badaniu ACTIVE była częstość występowania incydentów nowych złamań kręgosłupa u pacjentek leczonych abaloparatydem w porównaniu z placebo. W punkcie początkowym średni T-score wynosił -2,9 dla odcinka lędźwiowego kręgosłupa, -2,2 dla szyjki kości udowej oraz -1.9 dla całego stawu biodrowego. W punkcie początkowym 42% pacjentek nie doznało żadnych wcześniejszych złamań, 23% pacjentek przebyło co najmniej jedno wcześniejsze złamanie kręgów, a 43% pacjentek przebyło co najmniej jedno wcześniejsze złamanie inne niż złamanie kręgów.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Wpływ na występowanie nowych złamań kręgów W badaniu ACTIVE po 18 miesiącach abaloparatyd i teryparatyd znacząco zmniejszyły ryzyko bezwzględne nowych złamań kręgów w porównaniu z placebo u pacjentek w okresie pomenopauzalnym z osteoporozą (p < 0,0001; patrz tabela 2). Tabela 2 – Badanie ACTIVE: wpływ* abaloparatydu na ryzyko nowego złamania kręgów po 18 miesiącach
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Parametr PBO (N = 600) ABL (N = 583) TER (N = 600) Liczba pacjentek, u których wystąpiło złamaniekręgosłupa; n (%) 25 (4,2) 3 (0,5) 4 (0,7) Różnica w ryzyku bezwzględnym w porównaniu z placebo† (%)(95% CI) nie dotyczy 3,7 (2,0; 5,6) 3,5 (1,8; 5,5) - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
*Na podstawie zmodyfikowanej populacji wyodrębnionej zgodnie z zaplanowanym leczeniem (pacjentki, u których w punkcie początkowym i po rozpoczęciu leczenia wykonano radiogram kręgosłupa). † Różnicę w ryzyku bezwzględnym obliczano jako (PBO – ABL) i (PBO – TER). PBO = placebo, ABL = abaloparatyd, TER = teryparatyd, CI = przedział ufności Wpływ na występowanie nowych złamań innych niż złamania kręgów W badaniu ACTIVE po 19. miesiącach częstość występowania złamań innych niż złamania kręgów była podobna w grupie otrzymującej abaloparatyd (2,7%) i teryparatyd (2,0%), i nie różniła się statystycznie w porównaniu z placebo (3,6%) (patrz tabela 3). Tabela 3 – Badanie ACTIVE: czas do wystąpienia złamania innego niż złamanie kręgów po 19 miesiącach
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Parametr PBO (N = 688) ABL (N = 696) TER (N = 686) Częstość zdarzeń oszacowana na podstawie analizy Kaplana-Meiera (%)(95% CI) 3,6(2,3; 5,4) 2,7(1,6; 4,4) 2,0(1,1; 3,4) Liczba pacjentek, u których wystąpiło zdarzenien (%) 21 (3,1) 15 (2,2) 12 (1,7) Różnica w ryzyku bezwzględnym w porównaniu z placebo* (%)(95% CI) nie dotyczy 0,9(-1,1; 2,9) 1,6(-0,3; 3,5) - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
*Różnicę w ryzyku bezwzględnym obliczano jako (PBO – ABL) i (PBO – TER). PBO = placebo, ABL = abaloparatyd, TER = teryparatyd, CI = przedział ufności Wpływ na gęstość mineralną kości (ang. bone mineral density, BMD) W badaniu ACTIVE abaloparatyd znacząco zwiększał BMD we wszystkich mierzonych lokalizacjach anatomicznych w porównaniu z placebo po 6, 12 i 18 miesiącach. Średnia zmiana procentowa BMD w 18. miesiącu wynosiła 9,1% w porównaniu z 0,5% dla odcinka lędźwiowego kręgosłupa, 3,3% w porównaniu z 0% dla całego stawu biodrowego i 2,7% w porównaniu z -0,4% dla szyjki kości udowej odpowiednio w grupie otrzymującej abaloparatyd w porównaniu z grupą otrzymującą placebo (wszystkie p < 0,0001). Dla obszaru ultradystalnego kości promieniowej średnia zmiana procentowa BMD w 18. miesiącu wynosiła 1,2% w porównaniu z -1,0% w grupie otrzymującej abaloparatyd w porównaniu z grupą otrzymującą placebo.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
W grupie otrzymującej abaloparatyd wykazano stałe zwiększenie w pomiarach BMD, niezależnie od wieku, lat od menopauzy, rasy, regionu geograficznego, obecności lub braku obecności wcześniejszych złamań (kręgów lub innych niż kręgów), nasilenia choroby i BMD w punkcie początkowym. Markery obrotu kostnego U kobiet w okresie pomenopauzalnym z osteoporozą wykazano 90% zwiększenie stężenia markera kościotworzenia (s-PINP) po 1 miesiącu względem punktu początkowego i ten efekt utrzymywał się przez cały okres leczenia abaloparatydem. Nie wykazano zwiększenia stężenia markera resorpcji kostnej (s-CTX) po 1 miesiącu, ale wykazano przemijające 22% zwiększenie względem punktu początkowego po 3 miesiącach, po czym po zakończeniu leczenia wracało do wartości z punktu początkowego. Postępowanie po leczeniu Badanie przedłużone Po zakończeniu badania ACTIVE, 963 pacjentki zostały włączone do badania przedłużonego ACTIVExtend prowadzonego metodą otwartej próby, w którym wszystkie pacjentki otrzymywały leczenie 70 mg alendronianu (ALN) tygodniowo oraz suplementację wapnia i witaminy D do 24 miesięcy.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Tym badaniem objęto 494 pacjentki, które wcześniej otrzymywały placebo, i 469 pacjentek, które wcześniej otrzymywały abaloparatyd. Pacjentki, które w trakcie badania ACTIVE otrzymywały teryparatyd, nie kwalifikowały się do udziału w badaniu ACTIVExtend. Wyniki dotyczące zmniejszenia ryzyka złamania kręgów po 43 miesiącach od randomizacji przedstawiono w tabeli 4. Wpływ na występowanie nowych złamań kręgów – badanie przedłużone W badaniu ACTIVExtend po 43 miesiącach u pacjentek, które otrzymywały abaloparatyd/ALN, obserwowano znaczące zmniejszenie ryzyka bezwzględnego nowych złamań kręgów w porównaniu z pacjentkami, które otrzymywały placebo/ALN (p < 0,0001; patrz tabela 4). Nie badano leczenia alendronianem po stosowaniu teryparatydu. Tabela 4 – Badanie ACTIVExtend: wpływ* abaloparatydu/ALN na ryzyko nowego złamania kręgów po 43 miesiącach†
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Parametr PBO/ALN (N = 489) ABL/ALN (N = 457) Liczba pacjentek, u których wystąpiło złamaniekręgosłupa; n (%) 26 (5,3) 4 (0,9) Różnica w ryzyku bezwzględnym w porównaniu z placebo/ALN‡ (%)(95% CI) nie dotyczy 4,4(2,3; 6,9) - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
*Na podstawie zmodyfikowanej populacji wyodrębnionej zgodnie z zaplanowanym leczeniem (pacjentki, u których w punkcie początkowym i po rozpoczęciu leczenia wykonano radiogram kręgosłupa). † Początek leczenia alendronianem w 19. miesiącu ‡ Różnicę w ryzyku bezwzględnym obliczano jako (PBO/ALN – ABL/ALN). PBO = placebo, ABL = abaloparatyd, ALN = alendronian, CI = przedział ufności Wpływ na występowanie nowych złamań innych niż złamania kręgów – badanie przedłużone W badaniu ACTIVExtend po 43 miesiącach u pacjentek, które otrzymywały abaloparatyd/ALN, obserwowano liczbowe zmniejszenie ryzyka złamań innych niż złamania kręgów w porównaniu z pacjentkami, które otrzymywały placebo/ALN. Częstość występowania złamań innych niż złamania kręgów w grupie otrzymującej abaloparatyd/ALN (4,2%) nie była statystycznie istotna w porównaniu z grupą otrzymującą placebo (6,7%) (patrz tabela 5). Tabela 5 – Badanie ACTIVExtend: czas do wystąpienia złamania innego niż złamanie kręgów po 43 miesiącach*
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
Parametr PBO/ALN (N = 494) ABL/ALN (N = 469) Częstość zdarzeń oszacowana na podstawie analizy Kaplana-Meiera (%)(95% CI) 6,7(4,8; 9,3) 4,2(2,7; 6,4) Liczba pacjentek, u których wystąpiło zdarzenien (%) 32 (6,5) 19 (4,1) Różnica w ryzyku bezwzględnym w porównaniu z placebo/ALN† (%)(95% CI) nie dotyczy 2,5(-0,4; 5,4) - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakodynamiczne
* Początek leczenia alendronianem w 19. miesiącu † Różnicę w ryzyku bezwzględnym obliczano jako (PBO/ALN – ABL/ALN). PBO = placebo, ABL = abaloparatyd, ALN = alendronian, CI = przedział ufności Wpływ na gęstość mineralną kości (ang. bone mineral density, BMD) – badanie przedłużone Średnia zmiana procentowa BMD w 43. miesiącu wynosiła 14,7% w porównaniu z 6,8% dla odcinka lędźwiowego kręgosłupa, 6,3% w porównaniu z 2,9% dla całego stawu biodrowego, 5,0% w porównaniu z 1,6% dla szyjki kości udowej oraz 1,1% w porównaniu z 1,1% dla obszaru ultradystalnego kości promieniowej odpowiednio w grupie otrzymującej abaloparatyd/ALN w porównaniu z grupą otrzymującą placebo/ALN. Dzieci i młodzież Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań abaloparatydu we wszystkich podgrupach populacji dzieci i młodzieży w leczeniu osteoporozy (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Wchłanianie Po podaniu podskórnym mediana (zakres) czasu do osiągnięcia maksymalnego stężenia abaloparatydu w dawce 80 mikrogramów wynosiła 0,5 h (0,25 do 0,52 h). Bezwzględna biodostępność abaloparatydu u zdrowych uczestniczek po podaniu podskórnym dawki 80 mikrogramów wynosiła około 39%. Dystrybucja Wiązanie abaloparatydu z białkami osocza w warunkach in vitro wynosiło około 70%. Objętość dystrybucji wynosiła około 45 l. Metabolizm Nie przeprowadzono specyficznych badań dotyczących metabolizmu lub wydalania abaloparatydu. Metabolizm abaloparatydu polega na niespecyficznej proteolitycznej degradacji do mniejszych fragmentów peptydowych, po czym następuje eliminacja przez nerki. Badania in vitro wykazały, że abaloparatyd, w klinicznie istotnych stężeniach, nie hamuje ani nie indukuje enzymów cytochromu P450.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakokinetyczne
Eliminacja Średni pozorny całkowity klirens w osoczu po podaniu podskórnym wynosi 168 l/h u zdrowych pacjentek, a średni okres półtrwania abaloparatydu wynosi około 1 h. Fragmenty peptydów są eliminowane głównie przez nerki. Nie można wykluczyć czynnego wydzielania abaloparatydu przez nerki. Abaloparatyd nie jest substratem transporterów nerkowych P-gp, OAT1, OAT3, OCT2, MATE1 lub MATE2K. Ponadto abaloparatyd nie hamuje transporterów P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1 i OATP1B3 w warunkach in vitro w istotnych klinicznie stężeniach. Liniowość Ogólnie ekspozycja ogólnoustrojowa na abaloparatyd zwiększała się wraz ze zwiększaniem dawki podskórnej od 5 mikrogramów do maksymalnie 240 mikrogramów. Obserwowano ogólną tendencję do mniej niż proporcjonalnego do dawki wzrostu ekspozycji ogólnoustrojowej na abaloparatyd i nie obserwowano jej dalszego wzrostu po zwiększeniu dawki do 280 mikrogramów i 320 mikrogramów.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakokinetyczne
Zaburzenia czynności nerek Ekspozycja na abaloparatyd zwiększała się wraz ze zmniejszającym się CrCl. U pacjentek z łagodnymi, umiarkowanymi i ciężkimi zaburzeniami czynności wątroby występowało zwiększenie C max odpowiednio o 3%, 28% i 44% oraz zwiększenie AUC odpowiednio o 17%, 68% i 113% w porównaniu z pacjentkami z prawidłową czynnością nerek (patrz punkty 4.2 i 4.3). Nie przeprowadzono badań u pacjentek poddawanych dializie z powodu przewlekłej niewydolności nerek. Zaburzenia czynności wątroby Nie przeprowadzono badań u pacjentek z zaburzeniami czynności wątroby. Abaloparatyd jest peptydem, a nie inhibitorem bądź induktorem enzymów metabolizujących leki stosowane w chorobach wątroby. Eliminacja następuje poprzez proteolityczną degradację i wydalanie nerkowe, i jest mało prawdopodobne, aby zaburzenia czynności wątroby miały jakikolwiek istotny wpływ na ekspozycję na abaloparatyd. U tych pacjentek nie jest konieczna modyfikacja dawki (patrz punkt 4.2).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlWłaściwości farmakokinetyczne
Osoby w podeszłym wieku W badaniach klinicznych nie wykazano żadnych różnic w farmakokinetyce abaloparatydu zależnych od wieku, w tym u kobiet w okresie pomenopauzalnym w wieku od 49 do 86 lat.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie W trwającym 2 lata badaniu rakotwórczości na szczurach abaloparatyd prowadził do zwiększenia ogólnej częstości występowania kostniakomięsaka przy dawkach 4-krotnie większych niż ekspozycja ogólnoustrojowa obserwowana u ludzi po podaniu dawki podskórnej 80 mikrogramów, obliczanej przez porównania wartości AUC. Zmiany neoplastyczne związane z leczeniem abaloparatydem obejmowały zależne od dawki zwiększenie występowania kostniakomięsaków i kostniaków zarodkowych. Częstość występowania i najwcześniejsze wystąpienie guzów były podobne zarówno u samców, jak i samic szczurów. Znaczenie tych obserwacji dla człowieka jest nieznane, niemniej należy unikać stosowania abaloparatydu u pacjentek z grupy zwiększonego ryzyka wystąpienia kostniakomięsaka. W badaniach toksykologicznych na szczurach i małpach obserwowano mineralizację tkanek miękkich przy dawkach, odpowiednio, około 2- i 3-krotnie większych niż ekspozycja ogólnoustrojowa u ludzi w przypadku podawania podskórnego dawki 80 mikrogramów na dobę.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzedkliniczne dane o bezpieczeństwie
Podawanie podskórne abaloparatydu w dawkach około 0,3-, 2,4- i 3,8-krotnie większych niż ekspozycja u ludzi w przypadku podawania podskórnego dawki 80 mikrogramów na dobę przytomnemu psu prowadziło do zależnego od dawki przemijającego zwiększenia częstości akcji serca trwającego około 3 godzin; miało marginalny wpływ na średnie ciśnienie tętnicze krwi. Ponadto abaloparatyd miał marginalny wpływ na odstęp QTc, przy czym obserwowano nieistotną tendencję do zmniejszania odstępu QTc wraz ze zwiększaniem dawki, co jest spójne z minimalnym wpływem na prądy potasowe przepływające przez kanały jonowe typu hERG i włókna Purkinjego w klinicznie istotnych stężeniach. Abaloparatyd nie wykazywał działania genotoksycznego ani mutagennego w standardowym zestawie testów. Nie przeprowadzono badań dotyczących rozwoju zarodka i płodu ani rozwoju prenatalnego i poporodowego u samic zwierząt, ponieważ populacją docelową abaloparatydu są kobiety w okresie pomenopauzalnym.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlPrzedkliniczne dane o bezpieczeństwie
Wpływ na płodność samców oceniano u szczurów, i nie obserwowano żadnego wpływu na płodność samców przy dawkach 27-krotnie większych niż ekspozycja u ludzi w przypadku dawek podskórnych 80 mikrogramów na dobę.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Fenol Woda do wstrzykiwań Octan sodu trójwodny (do dostosowania pH) Kwas octowy (do dostosowania pH) 6.2 Niezgodności farmaceutyczne Nie mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności. 6.3 Okres ważności 3 lata Po pierwszym zastosowaniu lub po wyjęciu z lodówki wstrzykiwacz należy przechowywać w temperaturze poniżej 25 °C. Należy go użyć w ciągu 30 dni. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2 °C - 8 °C). Nie zamrażać. Warunki przechowywania po wyjęciu produktu leczniczego z lodówki, patrz punkt 6.3. 6.5 Rodzaj i zawartość opakowania Wkład (szkło silikonowane typu I) z tłokiem (guma chlorobutylowa), karbowanym wieczkiem (uszczelnienie z gumy chlorobutylowej)/aluminium umieszczony w jednorazowym wstrzykiwaczu. Każdy wstrzykiwacz półautomatyczny napełniony zawiera 1,5 ml roztworu (30 dawek).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Opakowanie zawierające 1 wstrzykiwacz półautomatyczny napełniony 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Każdy wstrzykiwacz powinien być używany wyłącznie przez jedną pacjentkę. Do każdego wstrzyknięcia musi być użyta nowa, jałowa igła. Wstrzykiwacz należy używać wyłącznie z igłami długości 8 mm i grubości 31 G. Do produktu leczniczego nie dołączono igieł. Wstrzykiwacza nie należy przechowywać z zamocowaną igłą. Produktu leczniczego Eladynos nie stosować, jeśli roztwór jest mętny, zabarwiony lub zawiera cząstki stałe. Przed rozpoczęciem stosowania wstrzykiwacza po raz pierwszy należy przeczytać i zrozumieć instrukcję dotyczącą używania wstrzykiwacza. Szczegółowa „Instrukcja użycia“ jest dołączona do pudełka ze wstrzykiwaczem. Wstrzykiwanie produktu leczniczego Eladynos
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Krok 1 Sprawdzenie wstrzykiwacza z produktem leczniczym Eladynoswstrzykiwacz. Krok 2 Zamocowanie igły do wstrzykiwacza z produktem leczniczym Eladynospodczas wprowadzania. Wstrzykiwacz nie będzie działać, jeśli igła nie będzie odpowiedniozamocowana. Nie dokręcać nadmiernie, ponieważ może to utrudnić usunięcie igły.wstrzyknięcia. Krok 3 Tylko w Dniu 1 – Testowanie wstrzykiwacza z lekiem Eladynos przed pierwszymwstrzyknięciemjednokrotnego przetestowania wstrzykiwacza, aby potwierdzić jego prawidłowe działanie.we wstrzykiwaczu szybko zabraknie leku. Dlatego Krok 3 należy wykonywać jeden raz dla każdego wstrzykiwacza, tylko w Dniu 1, przed pierwszym wstrzyknięciem.płyn w postaci kropli lub strumienia. Jeśli nie będzie widoczny żaden płyn, patrz„Rozwiązywanie problemów” w „Instrukcji użycia” na końcu ulotki dla pacjenta. Krok 4 Ustawienie dawki na wstrzykiwaczu z produktem leczniczym Eladynoswskaże „•80”. Wstrzykiwacz jest teraz gotowy do wykonania wstrzyknięcia. - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Przed użyciem wstrzykiwacza należy sprawdzić etykietę, aby upewnić się, że to właściwy Należy zanotować datę Dnia 1 w przeznaczonym do tego miejscu na pudełku. Wstrzykiwacza nie należy używać dłużej niż przez 30 kolejnych dni. Po 30 dniach od pierwszego użycia wstrzykiwacz należy usunąć. Zdjąć nasadkę z wstrzykiwacza. Sprawdzić wkład z produktem leczniczym Eladynos. Płyn powinien być przejrzysty, bezbarwny i wolny od cząstek stałych; w przeciwnym razie nie należy go stosować. Płyn może zawierać niewielkie pęcherzyki powietrza; jest to normalne. Usunąć papier ochronny z nowej strzykawki. Trzymając igłę z umieszczoną na nią nasadką prosto, nałożyć ją na wstrzykiwacz i przykręcić do oporu. Upewnić się, że igła jest trzymana prosto, aby nie wygięła się Zdjąć zewnętrzną nasadkę igły i zachować w celu ponownego użycia po wykonaniu Delikatnie zdjąć wewnętrzną nasadkę igły i usunąć.
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Wstrzykiwacz zawiera lek na 30 dni oraz niewielką dodatkową ilość w celu Uwaga: W przypadku testowania wstrzykiwacza przed każdym wstrzyknięciem Od Dnia 2 do Dnia 30 nie należy ponownie testować wstrzykiwacza, należy przejść bezpośrednio do Kroku 4 w celu ustawienia dawki do wstrzyknięcia. Należy przekręcić pokrętło ustawiania dawki na wstrzykiwaczu w kierunku od siebie (w kierunku zgodnym z ruchem wskazówek zegara), aż do oporu. Okienko wskazania dawki wskaże „•80” . Trzymać wstrzykiwacz igłą skierowaną ku górze. Nacisnąć zielony przycisk wstrzykiwania , aż do oporu. Na końcu igły będzie widoczny Okienko wskazania dawki wskaże „•0” . Należy przekręcić białe pokrętło na wstrzykiwaczu w kierunku od siebie (w kierunku zgodnym z ruchem wskazówek zegara), aż do oporu i aż okienko wskazania dawki
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Krok 5 Wybór i oczyszczenie miejsca wstrzyknięciaw czystą skórę. Nie wstrzykiwać w obszary na brzuchu, w których skóra jest delikatna, posiniaczona, zaczerwieniona, łuszcząca się lub twarda. Należy omijać obszary z bliznami lub rozstępami.i pozostawić do wyschnięcia. Krok 6 Podawanie wstrzyknięcia za pomocą wstrzykiwacza z lekiem EladynosWSZYSTKICH wymienionych poniżej zdarzeń i wyświetlenia wartości „•0”.ze skóry, a NASTĘPNIE zwolnić przycisk. Krok 7 Usuwanie igły ze wstrzykiwaczaz umieszczoną na niej nasadką odłączy się. Krok 8 Po wstrzyknięciuprzyłożyć bawełniany wacik lub gazik, aby zatrzymać krwawienie. Można również przykryć miejsce wstrzyknięcia małym plastrem samoprzylepnym. - CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Wstrzyknięcia należy podawać w podbrzusze. Należy unikać wstrzykiwania w promieniu 5 cm od pępka. Należy codziennie zmieniać miejsce wstrzyknięcia na brzuchu. Wstrzykiwać wyłącznie Należy przetrzeć miejsce wstrzyknięcia za pomocą wacika nasączonego alkoholem Po oczyszczeniu nie dotykać ani nie wachlować miejsca wstrzyknięcia, ani nie dmuchać na miejsce wstrzyknięcia. Uwaga: może być zalecane uchwycenie fałdu skóry w miejscu, w którym ma zostać wykonane wstrzyknięcie. Po wprowadzeniu igły w skórę rozluźnić uchwycony fałd skóry. Wprowadzić igłę w skórę pod kątem prostym. Przycisnąć i PRZYTRZYMAĆ zielony przycisk do momentu wystąpienia Należy przytrzymać przez 10 sekund , aby podać pełną dawkę, wysunąć wstrzykiwacz Ostrożnie umieścić zewnętrzną nasadkę igły z powrotem na igłę. Następnie ostrożnie wcisnąć zewnętrzną nasadkę igły, aż zaskoczy na miejsce, do oporu. Odkręcić igłę z umieszczoną na niej nasadką (tak jak odkręca się nakrętkę z butelki).
- CHPL leku Eladynos, roztwór do wstrzykiwań we wstrzykiwaczu, 2 mg/mlDane farmaceutyczne
Aby odkręcić igłę z umieszczoną na niej nasadką, należy ścisnąć nasadkę u podstawy, następnie przekręcić ją 8 lub więcej razy, po czym delikatnie pociągnąć, aż igła Uwaga: Podczas odkręcania igły nie naciskać na zewnętrzną nasadkę igły. Uwaga: Podczas odkręcania igły powinien być widoczny coraz większy odstęp między zewnętrzną nasadką igły a wstrzykiwaczem. Umieścić mocno nasadkę wstrzykiwacza z powrotem na wstrzykiwaczu. Pomiędzy wstrzyknięciami nasadkę należy pozostawić na wstrzykiwaczu z produktem leczniczym Eladynos. Może wystąpić niewielkie krwawienie, jest to normalne. Nie należy pocierać miejsca wstrzyknięcia. W przypadku wystąpienia niewielkiego krwawienia należy w razie potrzeby Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlNazwa produktu leczniczego, skład i postać farmaceutyczna
1. NAZWA PRODUKTU LECZNICZEGO EVENITY 105 mg roztwór do wstrzykiwań we wstrzykiwaczu półautomatycznym EVENITY 105 mg roztwór do wstrzykiwań w ampułko-strzykawce 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY EVENITY 105 mg roztwór do wstrzykiwań we wstrzykiwaczu półautomatycznym Każdy wstrzykiwacz półautomatyczny zawiera 105 mg romosozumabu w 1,17 ml roztworu (90 mg/ml). EVENITY 105 mg roztwór do wstrzykiwań w ampułko-strzykawce Każda ampułkostrzykawka zawiera 105 mg romosozumabu w 1,17 ml roztworu (90 mg/ml). Romosozumab to humanizowane przeciwciało monoklonalne IgG2 wytwarzane przy użyciu technologii rekombinacji DNA w komórkach jajnika chomika chińskiego (CHO). Substancje pomocnicze o znanym działaniu Każdy wstrzykiwacz półautomatyczny i każda ampułko-strzykawka zawiera 0,07 mg polisorbatu 20. Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Roztwór do wstrzykiwań (wstrzyknięcie). Roztwór klarowny do opalizującego, bezbarwny do bladożółtego.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWskazania do stosowania
4.1 Wskazania do stosowania Lek EVENITY jest wskazany do stosowania w leczeniu ciężkiej osteoporozy u kobiet po menopauzie z wysokim ryzykiem występowania złamań (patrz punkt 5.1).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDawkowanie
4.2 Dawkowanie i sposób podawania Leczenie należy rozpoczynać i nadzorować przez lekarzy specjalistów posiadająch doświadczenie w leczeniu osteoporozy. Dawkowanie Zalecana dawka to 210 mg romosozumabu (podawana w dwóch podskórnych wstrzyknięciach po 105 mg) raz w miesiącu przez 12 miesięcy. Pacjentki powinny otrzymywać wystarczające ilości wapnia i witaminy D przed i w trakcie leczenia (patrz punkty 4.3 i 4.4). Pacjentki leczone za pomocą leku EVENITY powinny otrzymać ulotkę dla pacjenta i kartę ostrzeżeń pacjenta. Po zakończeniu leczenia romosozumabem zalecane jest zastosowanie leczenia antyresorpcyjnego w celu utrzymania korzyści uzyskanych w leczeniu romosozumabem przez okres dłuższy niż 12 miesięcy. Pominięte dawki W przypadku pominięcia dawki romosozumabu należy ją podać tak szybko jak tylko możliwe. Kolejnej dawki romosozumabu nie należy przyjmować wcześniej niż jeden miesiąc po przyjęciu ostatniej dawki.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDawkowanie
Populacje szczególne Pacjentki w podeszłym wieku Dostosowywanie wielkości dawki u pacjentek w podeszłym wieku nie jest konieczne (patrz również punkt 5.2). Pacjentki z zaburzeniami czynności nerek Dostosowywanie wielkości dawki u pacjentek z zaburzeniami czynności nerek nie jest konieczne (patrz punkt 5.2). U pacjentek z ciężkimi zaburzeniami czynności nerek lub poddawanych dializie należy monitorować stężenie wapnia w surowicy krwi (patrz punkt 4.4). Pacjentki z zaburzeniami czynności wątroby Nie przeprowadzono żadnych badań klinicznych dotyczących wpływu na zaburzenia czynności wątroby (patrz punkt 5.2). Dzieci i młodzież Nie określono bezpieczeństwa stosowania ani skuteczności romosozumabu u dzieci i młodzieży (w wieku <18 lat). Dane nie są dostępne. Sposób podawania Podanie podskórne W celu podania dawki 210 mg należy wykonać 2 podskórne wstrzyknięcia romosozumabu w brzuch, udo lub górną część ramienia.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDawkowanie
Drugie wstrzyknięcie należy wykonać natychmiast po pierwszym, ale w innym miejscu podania. Lek powinna podawać osoba przeszkolona w sposobach wykonywania wstrzyknięć. Instrukcje dotyczące właściwego obchodzenia się z lekiem i jego usuwaniem przedstawiono w punkcie 6.6.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlPrzeciwwskazania
4.3 Przeciwwskazania - Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1 (patrz punkt 4.4) - Hipokalcemia (patrz punkt 4.4) - Przebyty zawał mięśnia sercowego lub udar mózgu (patrz punkt 4.4)
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Zawał mięśnia sercowego i udar mózgu W randomizowanych kontrolowanych badaniach, u pacjentów leczonych romosozumabem obserwowano wzrost liczby ciężkich zdarzeń naczyniowo-sercowych (zawału mięśnia sercowego i udaru mózgu) w porównaniu z grupą kontrolną (patrz punkt 4.8). Stosowanie romosozumabu jest przeciwwskazane u pacjentek, u których uprzednio wystąpił zawał mięśnia sercowego lub udar mózgu (patrz punkt 4.3). Podejmując decyzję o zastosowaniu romosozumabu u poszczególnych pacjentek, należy wziąć pod uwagę ryzyko wystąpienia złamań w kolejnym roku oraz ryzyko wystąpienia zdarzeń sercowo- naczyniowych na podstawie czynników ryzyka (np. zdiagnozowanej choroby sercowo-naczyniowej, nadciśnienia tętniczego, hiperlipidemii, cukrzycy, palenia tytoniu, ciężkiego zaburzenia czynności nerek, wieku).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
Romosozumab należy stosować wyłącznie w sytuacjach, w których lekarz przepisując produkt leczniczy i pacjntka zgadzają się, że korzyści przeważają nad ryzykiem. Jeśli u pacjentki wystąpi zawał mięśnia sercowego lub udar w trakcie leczenia, należy przerwać leczenie romosozumabem. Hipokalcemia U pacjentów otrzymujących romosozumab zaobserwowano występowanie przemijającej hipokalcemii. Przed rozpoczęciem leczenia romosozumabem należy skorygować hipokalcemię i pacjentki należy obserwować pod kątem oznak i objawów hipokalcemii. Jeżeli w trakcie leczenia u pacjentki pojawią się objawy sugerujące hipokalcemię (patrz punkt 4.8), należy zmierzyć stężenie wapnia. Pacjentki powinny otrzymywać wystarczające ilości wapnia i witaminy D (patrz punkty 4.3 i 4.8). U pacjentów z ciężkimi zaburzeniami czynności nerek (szacunkowy współczynnik przesączania kłębuszkowego [estimated glomerular filtration rate, eGFR] wynoszący 15 do 29 ml/min/1,73 m 2 ) lub poddawanych dializie ryzyko wystąpienia hipokalcemii jest większe, a dane dotyczące bezpieczeństwa stosowania w przypadku tych pacjentów są ograniczone.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
Należy monitorować stężenie wapnia u tych pacjentek. Nadwrażliwość W badaniach klinicznych w grupie pacjentów otrzymujących romosozumab występowały klinicznie istotne reakcje nadwrażliwości, w tym obrzęk naczynioruchowy, rumień wielopostaciowy oraz pokrzywka. W przypadku wystąpienia reakcji anafilaktycznej lub innej klinicznie istotnej reakcji alergicznej należy podjąć odpowiednie leczenie i przerwać stosowanie romosozumabu (patrz punkty 4.3 i 4.8). Martwica kości szczęki U pacjentów otrzymujących romosozumab przypadki wystąpienia martwicy kości szczęki (osteonecrosis of the jaw, ONJ) odnotowywano rzadko. Przy ocenianiu ryzyka wystąpienia u pacjentki ONJ należy uwzględniać następujące czynniki ryzyka: - siłę działania produktu leczniczego, który hamuje resorpcję kości (ryzyko zwiększa się wraz z siłą przeciwresorpcyjnego działania związku), oraz wielkość kumulatywnej dawki zastosowanej w terapii antyresorpcyjnej; - występowanie raka, współistnienie schorzeń (np.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
niedokrwistość, koagulopatia, zakażenia), palenie tytoniu; - terapie towarzyszące: kortykosteroidy, chemioterapia, inhibitory angiogenezy, radioterapia głowy i szyi; - niedostateczna higiena jamy ustnej, choroba przyzębia, źle dopasowane protezy, występowanie w przeszłości chorób zębów, inwazyjne zabiegi dentystyczne, np. usunięcie zębów. Wszystkie pacjentki powinny być zachęcane, aby w trakcie leczenia za pomocą romosozumabu utrzymywały dobrą higienę jamy ustnej, poddawały się rutynowym badaniom stomatologicznym i natychmiast zgłaszały wszelkie objawy występujące w jamie ustnej, takie jak ruchomość zębów, ból, obrzęk, niegojenie się ran lub obecność wydzieliny. Pacjentkom, w przypadku których istnieje podejrzenie o występowanie lub rozwijanie się ONJ w trakcie przyjmowania romosozumabu, należy zapewnić opiekę stomatologa lub chirurga szczękowego specjalizującego się w ONJ.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
Należy rozważyć przerwanie leczenia romosozumabem do czasu ustąpienia problemu i zminimalizowaniu przyczyniających się do niego czynników ryzyka tam, gdzie to możliwe. Atypowe złamania kości udowej U pacjentów otrzymujących romosozumab rzadko odnotowywano przypadki wystąpienia nietypowego niskoenergetycznego lub niskourazowego złamania trzonu kości udowej, które mogą występować spontanicznie. Jeżeli u pacjentki pojawi się nowy lub nietypowy ból w udzie, biodrze lub pachwinie, należy podejrzewać u niej atypowe złamanie i należy ją zbadać w celu sprawdzenia, czy nie doszło u niej do niepełnego złamania kości udowej. Pacjentki z atypowym złamaniem kości udowej należy również zbadać pod kątem objawów i oznak złamania drugiej kończyny dolnej. Należy rozważyć przerwanie leczenia romosozumabem, jeżeli wyniki oceny stosunku korzyści do ryzyka w danym przypadku za tym przemawiają.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlSpecjalne środki ostrozności
Substancje pomocnicze Ten produkt leczniczy zawiera 0,070 mg polisorbatu 20 w każdym wstrzykiwaczu półautomatycznym i w każdej ampułko-strzykawce. Polisorbaty mogą wywoływac reakcje alergiczne. Produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, to znaczy lek uznaje się się za „wolny od sodu”.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlInterakcje
4.5 Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji Nie przeprowadzono badań dotyczących interakcji romosozumabu z innymi produktami leczniczymi. Nie oczekuje się występowania interakcji farmakokinetycznych z romosozumabem.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWpływ na płodność, ciążę i laktację
4.6 Wpływ na płodność, ciążę i laktację Ciąża Romosozumab nie jest wskazany do stosowania u kobiet w wieku rozrodczym lub u kobiet w ciąży. Nie ma żadnych danych dotyczących stosowania romosozumabu u kobiet w ciąży. Tylko w jednym badaniu dotyczącym stosowania romosozumabu u szczurów rzadko odnotowano przypadki występowania wad rozwojowych w układzie kostnym (w tym syndaktylię i polidaktylię) (patrz punkt 5.3). Ryzyko wystąpienia wad rozwojowych w rozwijających się palcach ludzkiego płodu po ekspozycji na romosozumab jest małe w związku z tym, że u ludzi formowanie się palców ma miejsce w pierwszym trymestrze, w okresie gdy przenikanie immunoglobulin przez łożysko jest ograniczone. Karmienie piersią Romosozumab nie jest wskazany do stosowania u kobiet karmiących piersią. Nie ma żadnych danych dotyczących przenikania romosozumabu do mleka ludzkiego.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWpływ na płodność, ciążę i laktację
Wiadomo, że ludzkie immunoglobuliny G przenikają do mleka kobiet karmiących piersią w pierwszych kilku dniach po porodzie, po czym ich stężenie szybko maleje; w konsekwencji w tym krótkim okresie nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią. Płodność Brak jest danych dotyczących wpływu romosozumabu na płodność u ludzi. Badania na samicach i samcach szczurów nie wykazały żadnego wpływu na punkty końcowe dotyczące płodności (patrz punkt 5.3).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWpływ na zdolność prowadzenia pojazdów
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn Romosozumab nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
4.8 Działania niepożądane Zarys profilu bezpieczeństwa stosowania Najczęściej występującymi działaniami niepożądanymi były zapalenie nosogardzieli (13,6%) i bóle stawów (12,4%). Reakcje związane z nadwrażliwością wystąpiły u 6,7% pacjentów leczonych romosozumabem. Hipokalcemię odnotowywano niezbyt często (0,4% pacjentów leczonych romosozumabem). W randomizowanych, kontrolowanych badaniach u pacjentów leczonych romosozumabem obserowano wzrost liczby ciężkich zdarzeń sercowo-naczyniowych (zawałów serca i udarów mózgu) w porównaniu z grupą kontrolną (patrz punkt 4.4. oraz informacje podane poniżej). Tabelaryczny wykaz działań niepożądanych W klasyfikacji działań niepożądanych użyto następującej konwencji: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000) i bardzo rzadko (< 1/10 000).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
W obrębie każdej grupy o określonej częstości występowania oraz klasie układów i narządów działania niepożądane wymienione są zgodnie ze zmniejszającym się ich znaczeniem.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
Klasyfikacja układów i narządówMedDRA Działanie niepożądane Kategoria częstości Zakażenia i zarażenia pasożytnicze Zapalenie nosogardzieli Bardzo często Zapalenie zatok Często Zaburzenia układu immunologicznego NadwrażliwośćaWysypkaZapalenie skóry Często Pokrzywka Niezbyt często Obrzęk naczynioruchowy Rumień wielopostaciowy Rzadko Zaburzenia metabolizmu i odżywiania Hipokalcemiab Niezbyt często Zaburzenia układu nerwowego Ból głowy Często Udar mózguc Niezbyt często Zaburzenia oka Zaćma Niezbyt często Zaburzenia serca Zawał mięśnia sercowegoc Niezbyt często Zaburzenia mięśniowo-szkieletowe i tkanki łącznej Bóle stawów Bardzo często Ból szyi Skurcze mięśni Często Zaburzenia ogólne i stany w miejscupodania Reakcje w miejscu wkłuciad Często - CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
a. Patrz punkty 4.3 i 4.4. b. Definiowana jako niższe niż dolna granica normy stężenia wapnia w surowicy krwi korygowane o stężenie albumin. Patrz punkty 4.3 i 4.4. c. Patrz ustęp „Zawał mięśnia sercowego i udar mózgu i śmiertelność” poniżej. d. Najczęściej występującymi reakcjami w miejscu wkłucia były ból i rumień. Opis wybranych reakcji niepożądanych Immunogenność Odsetek kobiet po menopauzie przyjmujących co miesiąc romosozumab, u których występowały przeciwciała przeciwko romosozumabowi, wynosił 18,6% (1162 z 6244) w przypadku przeciwciał wiążących oraz 0,9% (58 z 6244) w przypadku przeciwciał neutralizujących. Przeciwciała przeciwko romosozumabowi zaczęły najwcześniej występować 3 miesiące po podaniu pierwszej dawki leku. Większość przypadków występowania przeciwciał była przemijająca. Obecność przeciwciał wiążących przeciwko romosozumabowi zmniejszała ekspozycję na lek o nawet 25%.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
W przypadkach obecności przeciwciał przeciwko romosozumabowi nie zaobserwowano żadnego wpływu na skuteczność leku. Z ograniczonych danych dotyczących bezpieczeństwa stosowania wynika, że odsetek występowania reakcji w miejscu wkłucia był ponadto liczbowo wyższy u pacjentek, u których występowały przeciwciała neutralizujące. Zawał mięśnia sercowego, udar mózgu i śmiertelność W badaniu dotyczącym stosowania romosozumabu w leczeniu ciężkiej osteoporozy u kobiet po menopauzie z grupą kontrolną otrzymującą aktywny komparator, w trakcie 12-miesięcznej fazy leczenia romosozumabem prowadzonej z zastosowaniem podwójnie ślepej próby, u 16 kobiet (0,8%) w grupie otrzymującej romosozumab wystąpił zawał mięśnia sercowego w porównaniu z 5 kobietami (0,2%) w grupie kontrolnej otrzymującej alendronian. Udar mózgu wystąpił u 13 kobiet (0,6%) w grupie otrzymującej romosozumab w porównaniu z 7 kobietami (0,3%) w grupie otrzymującej alendronian.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
Te zdarzenia wystąpiły u pacjentek z zawałem mięśnia sercowego w wywiadzie lub bez. Zgon z przyczyn sercowo-naczyniowych wystąpił u 17 kobiet (0,8%) w grupie otrzyjmującej romosozumab i u 12 kobiet (0,6%) w grupie otrzyjmującej alendronian. Liczba kobiet z ciężkimi niepożądanymi zdarzeniami sercowymi (orzeczony zgon z przyczyn sercowo-naczniowych, zawał mięśnia sercowego lub udar mózgu) wynosiła 41 (2,0%) w grupie otrzyjmującej romosozumab i u 22 (1,1%) w grupie otrzyjmującej alendronian, co daje współczynnik ryzyka na poziomie 1,87 (95% przedział ufności [1,11; 3,14]) dla romosozumabuu w porównaniu z alendronianem. Zgon z dowolnej przyczyny wystąpił u 30 kobiet (1,5%) w grupie otrzyjmującej romosozumab i u 22 kobiet (1,1%) w grupie otrzyjmującej alendronian. W badaniu dotyczącym stosowania romosozumabu w leczeniu osteoporozy u kobiet po menopauzie (w tym u kobiet z ciężką i mniej ciężką osteoporozą) z grupą kontrolną otrzymującą placebo, w trakcie 12- miesięcznej fazy leczenia romosozumabem prowadzonej z zastosowaniem podwójnie ślepej próby, nie było różnic w występowaniu ciężkich niepożądanych zdarzeń sercowych, 30 (0,8%) takich zdarzeń wystąpiło w grupie otrzyjmującej romosozumab i 29 (0,8%) w grupie otrzyjmującej placebo.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDziałania niepożądane
Zgon z dowolnej przyczyny wystąpił u 29 kobiet (0,8%) w grupie otrzyjmującej romosozumab i u 24 kobiet (0,7%) w grupie otrzyjmującej placebo. Zgłaszanie podejrzewanych działań niepożądanych Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V .
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlPrzedawkowanie
4.9 Przedawkowanie W badaniach klinicznych nie występowały przypadki przedawkowania. Nie jest znane żadne antidotum na romosozumab ani konkretne leczenie przedawkowania. W przypadku przedawkowania zaleca się uważne monitorowanie pacjentek i podjęcie odpowiedniego leczenia.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
5.1 Właściwości farmakodynamiczne Grupa farmakoterapeutyczna: leki stosowane w leczeniu chorób kości, leki wpływające na strukturę kości i mineralizację, kod ATC: M05BX06. Mechanizm działania Romosozumab to humanizowane przeciwciało monoklonalne (IgG2), które wiąże i hamuje aktywność sklerostyny, wzmagając w ten sposób proces kościotworzenia w związku z aktywowaniem komórek wyściółki kości, wzmagając wytwarzanie macierzy międzykomórkowej kości przez osteoblasty i rekrutowanie komórek osteoprogenitorowych. Ponadto romosozumab zmienia ekspresję mediatorów tworzenia osteoklastów, zmniejszając w ten sposób resorpcję kości. Rezultatem tego podwójnego efektu procesu kościotworzenia i zmniejszania resorpcji kości jest szybki wzrost w masie kości beleczkowatych i korowych oraz poprawa struktury i wytrzymałości kości.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Działania farmakodynamiczne U kobiet po menopauzie z osteoporozą stosowanie romosozumabu zwiększało na wczesnym etapie leczenia stężenie markera kościotwórczego, N-końcowego propeptydu prokolagenu typu I (P1NP), przy czym największy wzrost wynoszący 145% w stosunku do grupy otrzymującej placebo miał miejsce 2 tygodnie po rozpoczęciu leczenia, po czym w 9. miesiącu następował powrót do stężenia obserwowanego w grupie otrzymującej placebo, a następnie spadek w 12. miesiącu do około 15% poniżej poziomu resorpcji kości obserwowanego w grupie otrzymującej placebo. Stosowanie romosozumabu zmniejszało stężenie markera C-telopeptydu kolagenu typu I (CTX), zmniejszając go maksymalnie do około 55% w stosunku do poziomu obserwowanego w 2. tygodniu po rozpoczęciu leczenia w grupie otrzymującej placebo. Stężenie CTX pozostawało poniżej stężenia obserwowanego w grupie otrzymującej placebo i w 12. miesiącu było około 25% mniejsze od stężenia obserwowanego w grupie otrzymującej placebo.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Po zakończeniu leczenia romosozumabem u kobiet po menopauzie z osteoporozą stężenie P1NP wróciło do stężenia wyjściowego w ciągu 12 miesięcy; stężenie CTX wzrosło ponad stężenie wyjściowe w ciągu 3 miesięcy i wróciło do stężenia wyjściowego do 12. miesiąca, wskazując na odwracalność uzyskanych wyników. Po wznowieniu leczenia romosozumabem (u ograniczonej liczby pacjentów), po 12 miesiącach stosowania placebo, wzrost stężenia P1NP i spadek stężenia CTX wskutek stosowania romosozumabu były podobne do tych zaobserwowanych w czasie początkowego okresu leczenia. Skuteczność w badaniach klinicznych Leczenie osteoporozy u kobiet po menopauzie Skuteczność i bezpieczeństwo stosowania romosozumabu oceniane było w dwóch badaniach pilotażowych – badaniu z grupą kontrolną otrzymującą alendronian (ARCH) oraz badaniu z grupą kontrolną otrzymującą placebo (FRAME).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Badanie 20110142 (ARCH) Skuteczność i bezpieczeństwo stosowania romosozumabu w leczeniu osteoporozy u kobiet po menopauzie zostały poddane ocenie w badaniu wieloośrodkowym, międzynarodowym, randomizowanym, prowadzonym metodą podwójnie ślepej próby mającym na celu wykazanie wyższości badanego produktu leczniczego, z grupą kontrolną otrzymującą alendronian, z udziałem 4093 kobiet po menopauzie w wieku od 55 do 90 lat (średnia wieku 74,3 lat) z wcześniejszymi złamaniami z powodu kruchości kości. U kobiet włączonych do badania wskaźnik T dla mineralnej gęstości kości [Bone Mineral Density, BMD] w całym bliższym odcinku trzonowym kości udowej (ang. total hip) lub szyjki kości udowej wynosił ≤ −2,50 i występowało u nich przynajmniej 1 umiarkowane lub ciężkie złamanie kręgu lub przynajmniej 2 lekkie złamania kręgu lub też wskaźnik T dla BMD w całym bliższym odcinku trzonowym kości udowej lub szyjki kości udowej wynosił ≤ -2,00 i występowały u nich przynajmniej 2 umiarkowane lub ciężkie złamania kręgu lub złamanie proksymalnej części kości udowej, które nastąpiło w okresie od 3 do 24 miesięcy przed randomizacją.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Średnie wyjściowe wskaźniki T dla BMD odcinka lędźwiowego kręgosłupa, całego bliższego odcinka trzonowego kości udowej i szyjki kości udowej wynosiły 2,96; - 2,80 i - 2,90, odpowiednio, u 96,1% kobiet występowało złamanie kręgu w punkcie wyjściowym, a u 99,0% kobiet występowało wcześniejsze złamanie osteoporotyczne. Kobiety zostały zrandomizowane (w stosunku 1:1) do otrzymywania co miesiąc podskórnych wstrzyknięć romosozumabu lub przyjmowania co tydzień alendronianu w zaślepionej próbie przez 12 miesięcy. Po 12-miesięcznym okresie badania prowadzonym z zastosowaniem podwójnie ślepej próby kobiety w obu grupach przeszły na leczenie alendronianem, w dalszym ciągu nie znając szczegółów dotyczących początkowego okresu ich leczenia. Główna analiza została przeprowadzona, gdy wszystkie kobiety odbyły wizytę w 24. miesiącu badania. W jej wyniku potwierdzono wystąpienie złamań klinicznych u przynajmniej 330 kobiet, których mediana czasu wystąpienia przypadała po około 33.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
miesiącu obserwacji w badaniu. Kobiety otrzymywały codziennie suplementy wapnia i witaminy D. Pierwszorzędowymi punktami końcowymi skuteczności był odsetek występowania nowych złamań kręgu w okresie do 24. miesiąca oraz odsetek występowania złamań klinicznych (złamań innych niż złamania kręgu oraz złamań klinicznych kręgu) w czasie przeprowadzania głównej analizy. Wpływ na nowe złamania kręgu, złamania kliniczne, złamania inne niż złamania kręgu, złamania biodra i duże złamania osteoporotyczne Jak to pokazano w Tabeli 1, stosowanie romosozumabu zmniejszyło odsetek występowania nowych złamań kręgu w okresie do 24. miesiąca (skorygowana wartość p < 0,001) oraz odsetek występowania złamań klinicznych w czasie przeprowadzania głównej analizy (skorygowana wartość p < 0,001) oraz występowanie złamań innych niż złamania kręgu w głównej analizie (skorygowana wartość p = 0,040) w porównaniu z leczeniem samym alendronianem.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Tabela 1 pokazuje również zmniejszenie ryzyka wystąpienia złamań innych niż złamanie kręgu, złamań biodra i dużych złamań osteoporotycznych w czasie przeprowadzenia głównej analizy, do 12. i 24. miesiąca. Tabela 1. Wpływ stosowania romosozumabu na odsetek występowania i ryzyko wystąpienia nowych złamań kręgu, złamań klinicznych, złamań innych niż złamania kręgu, złamań biodra i dużych złamań osteoporotycznych u kobiet po menopauzie z osteoporozą
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Odsetek kobiet ze złamaniami Bezwzględne zmniejszenie ryzyka [%](95% CI (przedział ufności, ang. confidence interval)) Względne zmniejszenie ryzyka [%](95% CI) Alendronian/ alendronian (%) Romosozumab/ alendronian (%) Nowe złamania kręgu Do 12.miesiąca 85/1703 (5,0) 55/1696 (3,2) 1,84(0,51; 3,17) 36(11; 54) Do 24.miesiącaa 147/1834(8,0) 74/1825(4,1) 4,03(2,50; 5,57) 50(34; 62) Złamania kliniczneb Do 12.miesiąca 110/2047 (5,4) 79/2046 (3,9) 1,8(0,5; 3,1) 28(4; 46) Analiza główna (środkowy punkt czasowy okresu kontrolnego – ok. 33.miesiąc) 266/2047 (13,0) 198/2046 (9,7) NDc 27(12; 39) Złamania inne niż złamania kręgu Do 12miesiąca 95/2047 (4,6) 70/2046 (3,4) 1,4(0,1, 2,6) 26(-1, 46) Analiza główna (środkowy punkt czasowy okresu kontrolnego – ok. 33.miesiąc) 217/2047 (10,6) 178/2046 (8,7) NDc 19(1; 34) Złamania biodra Do 12.miesiąca 22/2047 (1,1) 14/2046 (0,7) 0,3(-0,3; 0,9) 36(-26; 67) Analiza główna (środkowy punkt czasowy okresu kontrolnego – ok. 33.miesiąc) 66/2047 (3,2) 41/2046 (2,0) NDc 38(8; 58) Duże złamania osteoporotyczned Do 12.miesiąca 85/2047 (4,2) 61/2046 (3,0) 1,4(0,3; 2,5) 28(-1; 48) - CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Odsetek kobiet ze złamaniami Bezwzględne zmniejszenie ryzyka [%](95% CI (przedział ufności, ang.confidence interval)) Względne zmniejszenie ryzyka [%](95% CI) Alendronian/ alendronian (%) Romosozumab/ alendronian (%) Analiza główna (środkowy punkt czasowy okresu kontrolnego – ok. 33.miesiąc) 209/2047 (10,2) 146/2046 (7,1) NDc 32(16; 45) - CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
a Bezwzględne i względne zmniejszenie ryzyka określone metodą Mantela-Haenszela z uwzględnieniem grupy wiekowej, wyjściowego wskaźnika T dla BMD w całym bliższym odcinku trzonowym kości udowej (ang. total hip) (≤ -2,5; > -2,5) oraz obecności ciężkich złamań kręgu w punkcie wyjściowym. Porównania pomiędzy grupami leczenia przeprowadzono przy użyciu skorygowanego modelu regresji logistycznej. b Złamania kliniczne obejmują wszystkie złamania objawowe, w tym złamania inne niż złamania kręgu oraz bolesne złamania kręgu. Porównania pomiędzy grupami leczenia przeprowadzono przy użyciu modelu proporcjonalnego ryzyka Coxa. c ND: nie dotyczy, ponieważ w czasie przeprowadzania głównej analizy okres ekspozycji uczestniczek na leki był różny. d Duże złamania osteoporotyczne obejmują złamania biodra, przedramienia, kości ramiennej oraz kliniczne złamania kręgu. Wpływ na gęstość mineralną kości (BMD) W 12. i 24.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
miesiącu stosowania romosozumabu u kobiet po menopauzie z osteoporozą przez 12 miesięcy po leczeniu alendronianem przez 12 miesięcy wartość BMD zwiększyła się w porównaniu z grupą otrzymującą alendronian (wartość p < 0,001) (patrz Tabela 2). Po 12 miesiącach leczenia z zastosowaniem romosozumabu, BMD w rejonie odcinka lędźwiowego kręgosłupa zwiększyło się w stosunku do punktu wyjściowego u 98% kobiet po menopauzie. Tabela 2. Średnia procentowa zmiana w BMD w okresie od punktu wyjściowego do 12. i 24. miesiąca u kobiet po menopauzie z osteoporozą
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Alendronian/AlendronianWartość średnia(95% CI)N = 2047a Romosozumab/AlendronianWartość średnia(95% CI)N = 2046a Różnica w wynikach leczenia w porównaniu z grupą kontynuującą leczeniealendronianem W 12. Miesiącu Lędźwiowy odcinek kręgosłupa 5,0(4,8; 5,2) 12,4 (12,1; 12,7) 7,4b (7,0; 7,8) Cały bliższy odcinek trzonowy kości udowej 2,9(2,7; 3,1) 5,8 (5,6; 6,1) 2,9b (2,7; 3,2) Szyjka kości udowej 2,0(1,8; 2,2) 4,9 (4,6; 5,1) 2,8b (2,5; 3,2) W 24. Miesiącu - CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Lędźwiowy odcinek kręgosłupa 7,2(6,9; 7,5) 14,0 (13,6; 14,4) 6,8b (6,4; 7,3) Cały bliższy odcinek trzonowy kości udowej 3,5(3,3; 3,7) 6,7 (6,4; 6,9) 3,2b (2,9; 3,6) Szyjka kości udowej 2,5(2,3; 2,8) 5,7 (5,4; 6,0) 3,2b (2,8; 3,5) - CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Wartości średnie i przedziały ufności dotyczą pacjentek, w przypadku których dostępne są dane. Na podstawie modelu ANCOVA brakująca zmiana w zakresie wartości podstawowego BMD i odsetka BMD w porównaniu z wartością w punkcie wyjściowym w miesiącu 12. i 24. została wprowadzona przy zastosowaniu imputaji statystycznej z grupą kontrolną. a Liczba zrandomizowanych kobiet b Wartość p < 0,001 Znaczna różnica w BMD zaobserwowana w pierwszych 12 miesiącach utrzymywała się po przejściu na leczenie alendronianem/kontynuowaniu leczenia alendronianem do 36. miesiąca. Różnice w wynikach leczenia w obrębie odcinka lędźwiowego kręgosłupa, całego bliższego odcinka trzonowego kości udowej i szyjki kości udowej widoczne były po 6 miesiącach. Badanie 20070337 (FRAME) Skuteczność i bezpieczeństwo stosowania romosozumabu w leczeniu osteoporozy pomenopauzalnej zostały poddane ocenie w badaniu wieloośrodkowym, międzynarodowym, randomizowanym, prowadzonym w grupach równoległych metodą podwójnie ślepej próby z grupą kontrolną otrzymującą placebo, z udziałem 7180 kobiet po menopauzie w wieku od 55 do 90 lat (średnia wieku 70,9 lat).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
U 40,8% kobiet włączonych do badania wystąpiła ciężka osteoporoza z wcześniejszym złamaniem w punkcie wyjściowym. Dodatkowymi głównymi punktami końcowymi skuteczności były odsetki występowania nowych złamań kręgu w okresie do 12. i 24. miesiąca. Stosowanie romosozumabu zmniejszyło odsetek występowania nowych złamań kręgu w okresie do 12. miesiąca (bezwzględne zmniejszenie ryzyka: 1,3% [95% CI: 0,79; 1,80], względne zmniejszenie ryzyka: 73% [95% CI: 53; 84], skorygowana wartość p < 0,001) i po przejściu na leczenie denosumabem do 24. miesiąca (bezwzględne zmniejszenie ryzyka: 1,89% [95% CI: 1,30; 2,49], względne zmniejszenie ryzyka: 75% [95% CI: 60; 84], skorygowana wartość p < 0,001). Kobiety przechodzące z leczenia bisfosfonianami Badanie 20080289 (STRUCTURE) Bezpieczeństwo stosowania i skuteczność romosozumabu było badane u kobiet po menopauzie z ciężką osteoporozą i przechodzących z leczenia bisfosfonianami (92,7% w grupie otrzymującej teryparatyd i 88,1% w grupie otrzymującej romosozumab przyjmowało wcześniej alendronian w ciągu ostatnich 3 lat) w badaniu wieloośrodkowym, randomizowanym, prowadzonym metodą otwartej próby w porównaniu do teryparatydu z udziałem 436 kobiet po menopauzie w wieku od 56 do 90 lat (średnia wieku 71,5 lat).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Pierwszorzędową zmienną skuteczności stosowania była procentowa zmiana ogólnej wartości wskaźnika BMD w całym bliższym odcinku trzonowym kości udowej w 12. miesiącu w porównaniu z wartością w punkcie początkowym. Romosuzumab istotnie zwiększał wartość wskaźnika BMD w całym bliższym odcinku trzonowym kości udowej w porównaniu do teriparatydu w 12. miesiącu (średnia różnica w leczeniu w porównaniu z teripartydem: 3,4% [95% CI: 2,8, 4,0], wartość p < 0,0001). Celem badania nie było sprawdzenie wpływu na występowanie złamań, lecz w grupie otrzymującej romosozumb wystąpiło siedem złamań, a w grupie otrzymującej teriparatyd – dziewięć. Histologia i histomorfometria kości W sub-badaniu związanym z histologią kości pobrano łącznie drogą biopsji 154 próbki z talerza kości biodrowej od 139 kobiet po menopauzie z osteoporozą w 2. i 12. miesiącu badania (badanie FRAME).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakodynamiczne
Histologiczne oceny jakościowe próbek wykazywały prawidłową architekturę i jakość kości we wszystkich punktach czasowych, prawidłową tkankę blaszkowatą bez obecności defektów mineralizacji, tkanki grubowłóknistej, zwłóknienia szpiku i klinicznie istotnych nieprawidłowości szpiku u pacjentek leczonych romosozumabem. Oceny histomorfometryczne bioptatów pobranych od kobiet w miesiącach 2. i 12. wykazywały zwiększenie się parametrów określających tworzenie się kości i zmniejszenie parametrów określających resorpcję kości wraz ze zwiększeniem się objętości kości i grubości kości beleczkowatych w grupie otrzymującej romosozumab, w porównaniu z grupą otrzymującą placebo. Dzieci i młodzież Europejska Agencja Leków wstrzymała obowiązek dołączania wyników badań romosozumabu w jednej lub kilku podgrupach populacji dzieci i młodzieży w leczeniu osteoporozy (stosowanie u dzieci i młodzieży: patrz punkt 4.2).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakokinetyczne
5.2 Właściwości farmakokinetyczne Wchłanianie Średni czas do osiągnięcia maksymalnego stężenia romosozumabu (t max ) wynosił 5 dni (zakres: 2 do 7 dni). Po podaniu podskórnie dawki 210 mg biodostępność wynosiła 81%. Metabolizm Romosozumab to humanizowane przeciwciało monoklonalne (IgG2) o wysokim powinowactwie i swoistości wobec sklerostyny, w związku z czym wydalany jest w drodze szybkiej wysycalnej eliminacji (tj. celowanego nieliniowego klirensu związanego z degradacją kompleksu romosozumab- sklerostyna) oraz w drodze powolnej nieswoistej eliminacji, w której pośredniczy układ siateczkowo- śródbłonkowy. Eliminacja Po osiągnięciu C max, stężenie w surowicy krwi opadało ze średnim efektywnym okresem półtrwania wynoszącym 12,8 dnia. Stan stacjonarny osiągany był na ogół do 3. miesiąca, przy czym akumulacja leku pozostawała na poziomie niższym niż 2-krotny po comiesięcznym podawaniu leku.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakokinetyczne
Liniowość lub nieliniowość Po podaniu podskórnym romosozumab wykazuje nieliniowe właściwości farmakokinetyczne w wyniku wiązania się ze sklerostyną. Wielkość podawanych dawek wahała się w granicach od 70 do 210 mg. Zaburzenia czynności nerek Po podaniu dawki 210 mg romosozumabu w badaniu klinicznym z udziałem 16 pacjentów z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny <30 ml/min) lub schyłkową niewydolnością nerek (end-stage renal disease, ESRD) poddawanych hemodializie średnie wartości C max i AUC były o 29% i 44% wyższe u pacjentów z ciężkimi zaburzeniami czynności nerek niż u zdrowych uczestników. Średnia ekspozycja na romosozumab była w przypadku pacjentów z ESRD poddawanych hemodializie podobna jak u zdrowych uczestników. Wyniki analizy farmakokinetycznej populacyjnej wskazywały na zwiększanie się ekspozycji na romosozumab ze zwiększającym się stopniem ciężkości zaburzeń czynności nerek.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakokinetyczne
Na podstawie modelu ekspozycja-odpowiedź dla zmian w BMD i porównania ze stopniem ekspozycji obserwowanym w przypadku tolerowanych klinicznie wielkościach dawki można jednak stwierdzić, że w przypadku tych pacjentów nie należy dostosowywać wielkości dawki. Zaleca się, aby pacjentki z ciężkimi zaburzeniami czynności nerek lub poddawanych dializie monitorować pod kątem hipokalcemii (patrz punkt 4.4). Zaburzenia czynności wątroby Nie przeprowadzono żadnych badań klinicznych dotyczących wpływu zaburzeń czynności wątroby. Nie oczekuje się, że występowanie zaburzeń czynności wątroby będzie miało wpływ na właściwości farmakokinetyczne romosozumabu, ponieważ wątroba nie jest głównym narządem uczestniczącym w metabolizmie lub wydalaniu romosozumabu. Pacjentki w podeszłym wieku Na właściwości farmakokinetyczne romosozumabu nie miał wpływu wiek w przedziale od 20 do 89 lat.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlWłaściwości farmakokinetyczne
Waga ciała Ekspozycja na romosozumab zmniejszała się wraz ze zwiększeniem masy ciała, jednak z analizy ekspozycja-odpowiedź wynika, że zmniejszenie to miało minimalny wpływ na wzrost BMD w obrębie odcinka lędźwiowego kręgosłupa i nie miało to klinicznego znaczenia. Z populacyjnych analiz PK wynika, że oczekiwana średnia wartość AUC w stanie stacjonarnym w przypadku pacjentów o wadze 61 kg i 114 kg wynosi odpowiednio 558 µg.doba/ml i 276 µg.doba/ml po comiesięcznym podawaniu podskórnie dawki 210 mg romosozumabu. Przynależność etniczna i płeć Nie jest konieczne dostosowywanie wielkości dawki ze względu na jakiekolwiek cechy osobowe pacjentki . Z populacyjnej analizy farmakokinetycznej wynika, że płeć i rasa (Japończycy w porównaniu z innymi rasami) nie miały żadnego klinicznego wpływu na właściwości farmakokinetyczne romosozumabu (zmiana w ekspozycji na lek w stanie stacjonarnym <20%).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlPrzedkliniczne dane o bezpieczeństwie
5.3 Przedkliniczne dane o bezpieczeństwie Dane niekliniczne dotyczące bezpieczeństwa stosowania, badań toksyczności po wielokrotnym podaniu oraz rakotwórczości, pochodzące z konwencjonalnych badań farmakologicznych lub badań kości dotyczących bezpieczeństwa, nie ujawniają szczególnego zagrożenia dla człowieka. W badaniu dotyczącym rakotwórczości podawano samcom i samicom szczurów Sprague-Dawley w wieku od 8 tygodni drogą wstrzyknięć podskórnych dawki o wielkości do 50 mg/kg mc./tydzień przez okres do 98 tygodni. Rezultatem podawania tych dawek była ogólnoustrojowa ekspozycja 19 razy wyższa niż ogólnoustrojowa ekspozycja obserwowana u ludzi po comiesięcznym podawaniu podskórnie dawki 210 mg romosozumabu (na podstawie porównań AUC). Romosozumab powodował zależny od dawki wzrost w masie kostnej przy makroskopowo widocznym pogrubieniu się kości we wszystkich dawkach. Nie zaobserwowano wpływu romosozumabu na śmiertelność ani na występowanie guzów u samców i samic szczurów.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlPrzedkliniczne dane o bezpieczeństwie
Badania na samicach i samcach szczurów nie wykazały żadnego wpływu przyjmowania romosozumabu na wyniki ocen dotyczących spółkowania, płodności lub zdolności reprodukcyjnych samców (parametrów dotyczących nasienia oraz wagi organów) i nie stwierdzono żadnego wpływu na cykl rujowy ani na jakiekolwiek parametry dotyczące jajników lub macicy przy ekspozycji na lek w dawkach około 54 razy wyższych niż stosowane w warunkach klinicznych. Rzadkie przypadki występowania wad rozwojowych w układzie kostnym, w tym syndaktylii i polidaktylii, odnotowano w przypadku 1 miotu na 75 przy ekspozycji na lek w dawkach około 30 razy wyższych niż stosowane w warunkach klinicznych, po podawaniu romosozumabu szczurom w okresie organogenezy. Nie stwierdzono żadnego niekorzystnego wpływu na wzrost i rozwój w okresie postnatalnym.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlPrzedkliniczne dane o bezpieczeństwie
Sugerowano, że sklerostyna odgrywa pewną rolę w formowaniu się palców, ponieważ jednak formowanie się palców u ludzi ma miejsce w pierwszym trymestrze, gdy przenikanie immunoglobulin przez łożysko jest ograniczone, ryzyko wystąpienia podobnych problemów u ludzi jest małe (patrz punkt 4.6).
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDane farmaceutyczne
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Wapnia octan Lodowaty kwas octowy Sodu wodorotlenek (w celu dostosowania pH) Sacharoza Polisorbat 20 Woda do wstrzykiwań 6.2 Niezgodności farmaceutyczne Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań zgodności. 6.3 Okres ważności 3 lata. Po wyjęciu leku EVENITY™ z lodówki w celu użycia nie należy go wkładać z powrotem do lodówki, ale można go przechowywać w temperaturze pokojowej (do 25°C) przez okres do 30 dni w oryginalnym opakowaniu. Jeżeli produkt nie zostanie wykorzystany w tym czasie, należy go wyrzucić. 6.4 Specjalne środki ostrożności podczas przechowywania Przechowywać w lodówce (2°C – 8°C). Nie zamrażać. Przechowywać ampułkostrzykawkę lub wstrzykiwacz półautomatyczny w opakowaniu zewnętrznym w celu ochrony przed światłem.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDane farmaceutyczne
6.5 Rodzaj i zawartość opakowania EVENITY 105 mg roztwór do wstrzykiwań we wstrzykiwaczu półautomatycznym Urządzenie przenośne do jednorazowego użytku do wykonywania wstrzyknięć z zamontowaną ampułko-strzykawką zawierającą 1,17 ml roztworu. Strzykawka wewnątrz wstrzykiwacza wykonana jest z polimeru cykloolefinowego i wyposażona w korek (chlorobutylowy) oraz formowaną igłę ze stali nierdzewnej z osłoną igły z elastomeru (gumy syntetycznej). Opakowanie zawiera 2 wstrzykiwacze półautomatyczne. Wielopak zawierający 6 (3 opakowania po 2) wstrzykiwaczy półautomatycznych. EVENITY 105 mg w postaci roztworu do wstrzykiwań w ampułko-strzykawce Ampułko-strzykawka do jednorazowego użytku zawierająca 1,17 ml roztworu. Strzykawka wykonana jest z polimeru cykloolefinowego i wyposażona w korek (chlorobutylowy) oraz formowaną igłę ze stali nierdzewnej i osłonę igły z elastomeru (gumy syntetycznej). Opakowanie zawiera 2 ampułko-strzykawki. Wielopak zawierający 6 (3 opakowania po 2) ampułko-strzykawek.
- CHPL leku Evenity, roztwór do wstrzykiwań we wstrzykiwaczu, 90 mg/mlDane farmaceutyczne
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie. 6.6 Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania Przed podaniem roztwór należy sprawdzić wizualnie pod kątem obecności cząsteczek stałych i przebarwienia. Leku EVENITY nie należy używać, jeżeli roztwór jest przebarwiony, mętny lub zawiera cząsteczki stałe. Przed podaniem podskórnym należy romosozumab pozostawić w temperaturze pokojowej przez co najmniej 30 minut przed jego wstrzyknięciem. Dzięki temu podanie będzie bardziej komfortowe. Nie należy go ogrzewać w żaden inny sposób. Nie wstrząsać. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Zobacz również:
- Dawkowanie leku
Teryparatyd to nowoczesna substancja czynna stosowana głównie w leczeniu osteoporozy u dorosłych, która może znacząco zmniejszyć ryzyko złamań kości. Jego dawkowanie jest jasno określone, ale wymaga ścisłego przestrzegania zaleceń, zwłaszcza w określonych grupach pacjentów, takich jak osoby starsze czy z zaburzeniami pracy nerek. Dowiedz się, jakie są standardowe schematy podawania teryparatydu i na co warto zwrócić uwagę w trakcie leczenia.
- Działania niepożądane i skutki uboczne
Teryparatyd to substancja czynna stosowana głównie w leczeniu osteoporozy oraz niektórych zaburzeń gospodarki wapniowo-fosforanowej. Jak każdy lek, może wywoływać działania niepożądane, które najczęściej mają łagodny lub umiarkowany charakter, ale czasami bywają poważniejsze. W zależności od postaci leku, drogi podania i indywidualnych cech pacjenta, profile działań niepożądanych mogą się nieco różnić. Poznaj najważniejsze możliwe skutki uboczne stosowania teryparatydu oraz sposoby ich rozpoznania.
- Mechanizm działania
Mechanizm działania teryparatydu opiera się na pobudzaniu naturalnych procesów tworzenia kości w organizmie. Substancja ta działa na poziomie komórkowym, aktywując komórki odpowiedzialne za budowę kości oraz regulując gospodarkę wapniową. Dzięki temu teryparatyd pomaga w leczeniu chorób takich jak osteoporoza czy przewlekła niedoczynność przytarczyc, przyczyniając się do poprawy wytrzymałości kości i zmniejszenia ryzyka złamań.
- Porównanie substancji czynnych
Teryparatyd, abaloparatyd oraz romosozumab należą do grupy leków stosowanych w leczeniu osteoporozy, ale różnią się pod względem wskazań, mechanizmu działania i bezpieczeństwa stosowania. Poznaj podobieństwa i kluczowe różnice między tymi substancjami, aby lepiej zrozumieć, kiedy i dlaczego są wybierane w terapii wzmacniania kości.
- Profil bezpieczeństwa
Teryparatyd to substancja czynna stosowana głównie w leczeniu osteoporozy i niedoczynności przytarczyc. Chociaż jego skuteczność została potwierdzona w licznych badaniach, bezpieczeństwo stosowania zależy od wielu czynników, takich jak wiek pacjenta, stan nerek czy wątroby oraz towarzyszące choroby. W niniejszym opisie znajdziesz najważniejsze informacje dotyczące bezpiecznego stosowania teryparatydu, w tym zalecenia dla kobiet w ciąży i karmiących piersią, wpływ na prowadzenie pojazdów, interakcje z innymi lekami i alkoholem oraz wskazówki dotyczące stosowania u osób starszych i w szczególnych grupach pacjentów.
- Przeciwwskazania
Teryparatyd to substancja czynna wykorzystywana w leczeniu osteoporozy, która skutecznie wspiera odbudowę kości i zmniejsza ryzyko złamań u osób z grupy podwyższonego ryzyka. Jednak nie każdy pacjent może ją stosować – istnieją konkretne przeciwwskazania, które należy wziąć pod uwagę przed rozpoczęciem terapii. Poznaj szczegółowo, kiedy teryparatyd jest niewskazany, w jakich przypadkach należy zachować ostrożność oraz co warto wiedzieć, aby terapia była bezpieczna.
- Stosowanie u dzieci
Bezpieczeństwo stosowania teryparatydu u dzieci jest zagadnieniem szczególnie istotnym ze względu na różnice w funkcjonowaniu organizmu dzieci i dorosłych. Choć teryparatyd odgrywa ważną rolę w leczeniu osteoporozy u dorosłych, jego stosowanie w populacji pediatrycznej nie jest zalecane. Dowiedz się, dlaczego dzieci wymagają szczególnej ostrożności przy podawaniu leków oraz jakie są oficjalne zalecenia dotyczące teryparatydu.
- Stosowanie u kierowców
Teryparatyd jest nowoczesną substancją czynną stosowaną głównie w leczeniu osteoporozy i zaburzeń gospodarki wapniowo-fosforanowej. Choć ogólnie nie wpływa znacząco na zdolność prowadzenia pojazdów i obsługi maszyn, u niektórych osób może wywołać przemijające zawroty głowy czy spadki ciśnienia. Poznaj, na co warto zwrócić uwagę podczas terapii teryparatydem, aby zachować bezpieczeństwo w codziennym życiu.
- Stosowanie w ciąży
Stosowanie leków w ciąży i podczas karmienia piersią wymaga wyjątkowej ostrożności, ponieważ wiele substancji może wpływać na zdrowie dziecka. Teryparatyd, wykorzystywany głównie w leczeniu osteoporozy, to lek, którego bezpieczeństwo w tych szczególnych okresach życia kobiety jest bardzo jasno określone w dokumentacji medycznej. Poznaj, jakie są zalecenia dotyczące teryparatydu w ciąży, podczas karmienia piersią oraz co wiemy o jego wpływie na płodność.
- Wskazania - na co działa?
Teryparatyd to nowoczesna substancja czynna, która znajduje zastosowanie w leczeniu poważnych schorzeń związanych z układem kostnym oraz zaburzeniami gospodarki wapniowo-fosforanowej. Dzięki swojemu unikalnemu działaniu pobudza procesy budowy kości i może znacząco zmniejszać ryzyko złamań u pacjentów dorosłych z osteoporozą, a także wspierać leczenie przewlekłej niedoczynności przytarczyc. Dowiedz się, dla kogo teryparatyd jest wskazany, jak działa i w jakich sytuacjach przynosi największe korzyści.
- Rzedawkowanie substancji
Teryparatyd to substancja wykorzystywana w leczeniu osteoporozy i zaburzeń gospodarki wapniowej. Przedawkowanie tej substancji może prowadzić do poważnych objawów, takich jak zaburzenia poziomu wapnia we krwi, nudności, osłabienie czy zawroty głowy. W opisie znajdziesz szczegółowe informacje na temat objawów przedawkowania teryparatydu, możliwych konsekwencji oraz zalecanego postępowania w takich sytuacjach, z uwzględnieniem różnych postaci i dróg podania tej substancji.
REKLAMA